


1. Background
T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive form of blood cancer. It is a type of a broader category of leukemia called acute lymphoblastic leukemia (ALL). T-cell acute lymphoblastic leukemia accounts for approximately 12% - 15% of ALL cases in children and nearly 25% of cases in adults (1). One of the targets to limit the proliferation of these cells is the inhibition of inosine monophosphate dehydrogenase by reducing the synthesis of DNA nucleotides (2). Inosine monophosphate dehydrogenase (IMPDH) is one of the most important enzymes in the life cycle of lymphocytes, catalyzing the NAD-dependent conversion of inosine 5’-monophosphate to xanthine monophosphate, which is the rate-limiting step in de novo guanine nucleotide biosynthesis (3). The proliferation of T- and B-lymphocytes mainly depends on the presence of a large number of guanine nucleotides. Therefore, the enzyme is upregulated in these cells to support this activity (4).
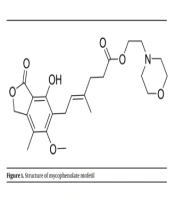
Lymphocytes cannot use the salvage pathway to provide purines for their life cycle, and they only rely on the de novo synthesis of purines. Consequently, inhibiting IMPDH is one of the best choices to suppress their proliferation. The synthesis of enzyme antagonists is a subject of many studies on treating immune system disorders, such as graft-versus-host disease (GvHD) and cancers (5). One of the well-known active ingredients that inhibit IMPDH is mycophenolic acid, which is found in the formulation of various medicines, such as mycophenolate mofetil (Figure 1).

Structure of mycophenolate mofetil
This medicine is commonly used as immunosuppressive in the prophylaxis of allograft rejection in transplant recipients (6-8) and autoimmune diseases, such as Crohn’s (9), lupus (10), and anticancer agents (11). Mycophenolic acid reduces the guanosine triphosphate (GTP) pool resulting in a cycle arrest mainly in G0/G1 phase, although some blockage in the S phase has also been reported (12, 13). However, a high dose of MPA, more than 1 g/day, is needed to prevent transplant rejection in patients, and this dose might cause many serious side effects, including anemia (14), gastrointestinal bleeding (15), progressive multifocal leukoencephalopathy (16), and flashing.
Proteolysis-targeting chimera is a bifunctional molecule with two attached parts via a linker, the head has a ligand to recognize the targeted protein to be degraded, and the other has an E3 ligase ligand (17). The proximity of the protein and E3 ligase can induce the ubiquitination of the protein in lysine, serine, or threonine residues (18) and leads to degradation through the ubiquitin-proteasome system (UPS). After that, the PROTAC is intact and ready to target another protein. Therefore, in a sub-stoichiometric amount, a biological response is achieved with less total drug concentration (17). Selectivity, longer-acting activity, and less off-target side effects of this technology in comparison with the traditional inhibition of proteins make it a promising approach to drug discovery (19). Since the first introduction by Crews and coworkers in 2001, the PROTACs have been broadly applied for degrading various proteins, such as Sirtuin 2 (20), bromodomain and extra-terminal (BET) (21), TANK-binding kinase 1 (TBK1) (22), B-cell lymphoma 6 (BCL6) (23), and estrogen-related receptor α (24). To use the advantages of the PROTAC technique, we have synthesized novel compounds to degrade the IMPDH by the proteasome system. By this means, alkynylated MPA was coupled with pomalidomide-derived azide via click reaction. In other compounds, MPA and pomalidomide-derived azides were synthesized and attached by different lengths of alkynylated glycol linkers through a copper-catalyzed click cycloaddition reaction. The versatile ligands were chosen to strengthen the interaction and minimize the proximity of E3 ligase and IMPDH (Figure 2).

Proteolysis-targeting chimera derivatives
On the other hand, the presence of triazole moiety in the framework of a molecule generally provides some advantages, including increasing water solubility, strengthening interaction with receptors through the addition of hydrogen bond acceptor to structure, raising the rigidity of PROTACs to fix the molecule in a receptor (20), and broad functional groups tolerance of click chemistry in the synthesis of PROTACs.
2. Objectives
In this study, the Jurkat cell line was chosen as a model of acute T-cell leukemia to evaluate the activity of compounds against the proliferation of these cells as anti-leukemia agents.
3. Methods
3.1. Materials and Instruments
All reagents were obtained from commercial suppliers and were used without further purification. Semi-preparative high-performance liquid chromatography (HPLC) purification was performed on a Knauer UV-directed purification system equipped with an 1800 Binary Gradient Module and 15 × 25 mm C18 column. The mobile phases were water ((0.1% trifluoroacetic acid (TFA)) and acetonitrile with a flow rate of 10 mL/min. The semi-preparative purification gradient was 100% water for 5 min and was run to 100% acetonitrile in 55 min. In order to change the salt of the purified compound from TFA to acetate, the mobile phases were water (1% acetic acid) and acetonitrile with a flow rate of 10 mL/min, and the same gradient was applied as described. 1H NMR (400 MHz) and 13C NMR (100.6 MHz) spectra were recorded on Bruker spectrometers. The purity of all final compounds was over 95% determined by analytical HPLC at 220 nM.
3.2. Chemistry
The molecular structure of PROTAC needs a protein-targeting constituent connected via a linker to an E3 ligase ligand. To find the best distance, pomalidomide and four derivatives of MPA with different linkers were synthesized and purified by semi-preparative HPLC (Figure 3). According to Figure 3, for the synthesis of products 1 and 2, compounds 5 (25) and 6 (26) reacted with MPA in the presence of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU) as a coupling reagent to obtain 7 and 8, respectively. Compounds 5 and 6 can be synthesized from the reaction of Boc-ethanolamine and Boc-propanolamine with propargyl bromide, respectively, followed by a deprotection process using TFA in dichloromethane (25, 26).
-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU), DIEA, r.t, 4 h, 42% yield; B, THF, H<sub>2</sub>O, 9, CuSO<sub>4</sub>.5H<sub>2</sub>O, sodium ascorbate, r.t, 12 h, 47% yield; C, DMF, TBTU, DIEA, r.t, 4 h, 56% yield; D, THF, H<sub>2</sub>O, 12 or 13, 4 eq, CuSO<sub>4</sub>.5H<sub>2</sub>O, sodium ascorbate, r.t, 12 h, 60% yield; E, THF, H<sub>2</sub>O, CuSO<sub>4</sub>.5H<sub>2</sub>O, sodium ascorbate, r.t, 12 h, 63% yield")
Reagents and conditions: A, DMF, 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU), DIEA, r.t, 4 h, 42% yield; B, THF, H2O, 9, CuSO4.5H2O, sodium ascorbate, r.t, 12 h, 47% yield; C, DMF, TBTU, DIEA, r.t, 4 h, 56% yield; D, THF, H2O, 12 or 13, 4 eq, CuSO4.5H2O, sodium ascorbate, r.t, 12 h, 60% yield; E, THF, H2O, CuSO4.5H2O, sodium ascorbate, r.t, 12 h, 63% yield
In the next step, compound 9 (27) was first synthesized from the reaction of pomalidomide with 2-bromoacetic acid and sodium azide. Next, it reacted with 7 and 8 in the presence of CuSO4.5H2O and sodium ascorbate for the in situ production of Cu(I), furnishing the desired products 1 and 2, respectively. After the filtration of the reaction mixture through a plug of celite and evaporation of the solvent, the residue was purified using semi-prep HPLC with 0.1% TFA as the mobile phase A and acetonitrile as the mobile phase B.
Trifluoroacetic acid must be exchanged with an acetate ion due to toxicity to living cells. To this aim, the purified compounds were reinjected to semi-prep HPLC and were washed with 1% acetic acid as the mobile phase A and acetonitrile as the mobile phase B. Finally, the pure fractions were merged and freeze-dried. In the synthetic routes of products 3 and 4, compound 10 (28) was initially synthesized starting from the reaction of 3-bromopropylamine with sodium azide. Afterwards, it reacted with MPA in the presence of TBTU as a coupling reagent to give 11. Correspondingly, compounds 12 and 13 (29) were synthesized from the reaction of diethylene glycol and triethylene glycol with propargyl bromide, respectively. Then, they reacted with 11 through a Cu(I)-catalyzed 1,3-dipolar cycloaddition reaction to obtain adducts 14 and 15, respectively. Following the purification of these products by semi-preparative HPLC and freeze-drying, in the final step of preparing products 3 and 4, they reacted with 9 through click reaction. After filtrating the reaction mixture using celite and evaporating the solvent, the residue was purified and ion-exchanged, as aforementioned.
3.3. Biological Evaluation
To evaluate the potency of PROTACs against the Jurkat cell line as immortalized T lymphocyte cells, the Annexin V-FITC and Western blot tests were performed.
3.3.1. Flow Cytometry
Apoptosis was assessed by Annexin V and PI double staining of treated and non-treated Jurkat cells using the PADZA apoptosis kit following the manufacturer’s protocol. In brief, the Jurkat cell line was cultured in DMEM/F12 supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum. The cells were incubated at 37°C and 5% CO2. For apoptosis assay, the cells were seeded in 96-well plates at 1 × 105 cells /mL (1 × 104 cells/100 μL or well) and were incubated at 37°C and 5% CO2 for 24 h. Subsequently, the cells were treated with different dilutions of the compounds in each well in triplicates for 24 h at 37°C. After incubation, the microplates were centrifuged at 250 g for 5 min. Next, cells were washed with cold phosphate-buffered saline (PBS) and resuspended in 200 μL of binding buffer 1X, followed by incubation with 2 μL of Annexin V-FITC for 20 min at room temperature in the dark. Then, cells were washed using a binding buffer, resuspended in 200 μL of binding buffer 1X supplemented with 2 μL of PI, and then analyzed using a flow cytometer within 1 h. In our study, apoptotic cells were in the early (Annexin V+, PI-) and late (Annexin V+, PI+) apoptosis stage, while viable cells were negative for both Annexin V and PI. As a positive control for apoptosis, Jurkat cells were treated with staurosporine for 5 h. By necrosis induction, the cells were boiled in culture for 1 min.
3.3.2. Immunoblotting Analysis
The western blot technique was performed to determine the cytoplasmic level of IMPDH. Cells were harvested and treated with different doses of compound 2 alone and with bortezomib (BTZ) for 24 h. Briefly, cells were homogenized in ice-cooled RIPA lysis buffer (50 mM Tris-HCl (pH 8.0), 0.1% sodium dodecyl sulfate, 150 mM sodium chloride, 0.5% sodium deoxycholate, and 1.0% NP-40, and were then centrifuged at 12,000 g for 20 min at 4°C. The supernatant was collected, and protein concentration was determined using NanoDrop Spectrophotometer (Thermo Fisher Scientific). A 10% SDS-polyacrylamide gel was used to separate the proteins by electrophoresis, and proteins were transferred to the methanol pre-activated polyvinylidene fluoride membranes. To block nonspecific binding, membranes were incubated in blocking bovine solution serum accepted manuscript albumin 1% in phosphate-buffered saline plus 0.1% Tween-20 for 2 h with a gentle shake. Subsequently, blots were incubated with different primary rabbit antibodies anti-IMPDH: sc-166551 (Santa Cruz, USA, 1: 500) and anti-β-actin sc-47778 (1: 300) overnight at 4°C. After four times washing with PBS, the blots were finally incubated with horseradish peroxidase (HRP) conjugated anti-mouse secondary antibody (m-IgGκBP-HRP: sc-516102; Santa Cruz, USA, 1: 5000) for 1 h at room temperature. The protein band was visualized by the enhanced chemiluminescence method. In this study, β-actin was used as an internal loading control.
4. Results
Due to the IMPDH role in purine synthesis, especially in lymphocyte cells that only rely on the de-novo synthesis of purines, the inhibition of this enzyme causes extreme deficiency of purines, such as ATP and guanine, leading to energy distress and DNA replication disorders (30). As a result, all these consequences directed lymphocytes to apoptosis. The test was determined by Annexin V and PI double staining of treated and non-treated Jurkat cells using the PADZA apoptosis kit. As shown in Figure 4, in the apoptosis assay with Annexin V-FITC, all compounds (almost 50 nM) induced 86% and 14% apoptosis in the early and late phases, respectively.

A, Apoptosis plot of derivatives and MPA at 50 nM; B, Necrosis-induced Jurkat cell; C, Apoptosis-induced Jurkat cell; D, Untreated Jurkat cell
The apoptosis test was also accomplished at 0.1, 1, and 10 μM, and the results showed similar data in ESI. Regardless of compound concentrations, any shortage of purines pool in lymphocyte cancer cells causes apoptosis. Therefore, it is not a dose-dependent response. To verify the validity of the PADZA kit, necrosis, and apoptosis Jurkat cells were prepared by boiling in cell culture and treated with staurosporine, respectively.
To prove the concept of MPA-based compound 2 for chemically-induced IMPDH degradation, the cytoplasmic level of IMPDH in Jurkat cells was measured after incubation with 2 via western blot analysis. As shown in Figure 5A, the intensity of the IMPDH bond decreased by increasing the dose of 2 from 0.05 μM to 0.5 μM. According to Figure 5B, the quantity of IMPDH in treated cells significantly declined to 27% and 10% of untreated Jurkat cells in 0.05 μM to 0.5 μM, respectively. It seems that the proteasomal degradation of IMPDH stopped at 10 μM because the ubiquitin-proteasome system was saturated. A reliable way to prove the proteasomal degradation of proteins is using potent proteasome inhibitors, such as BTZ. We could show that the effect of 2 in 0.05 μM on IMPDH degradation can be almost prevented by competition with BTZ as the proteasome inhibitor in 0.1 and 0.5 μM. In comparison to untreated cells, the results demonstrated that adding BTZ at 0.1 and 0.5 μM can raise the IMPDH quantity by 70% and 84%, respectively. On the other hand, treatment of Jurkat cells with MPA does not affect the cytoplasmic level of IMPDH. Consequently, all these observations indicate the proteasomal degradation of IMPDH by our novel molecules.

Western blot analysis
5. Discussion
In the current study, we reported the novel structure-based development of a PROTAC for the IMPDH enzyme. The PROTACs molecules offer several advantages compared to traditional enzyme inhibitors, especially their selectivity and potency. Therefore, our novel tool for IMPDH degradation will open up a new roadmap to eliminate the drawbacks and side effects. Meanwhile, through the reaction between azido-pomalidomide and alkynylated glycols, some novel compounds were synthesized that are ready to be “clicked” to other ligands, furnishing new PROTACs.
5.1. Experiment
The detailed experimental procedures, NMR data (e.g., 1H NMR and 13C NMR spectra), mass data, analytical HPLC chromatogram, and quantitative western blot data are all provided in the supporting information.
