











1. Background
Epidemiological and animal studies indicate that an adverse intrauterine environment is associated with an increased risk of cardiovascular diseases in adulthood (1). Prenatal hypoxia, the most common adverse intrauterine environment, can make the fetus unable to realize its genetically-determined growth potential, manifested as growth retardation and organ function injury in offspring (2). Chronic fetal hypoxia often results from pregnancy with increased placental vascular resistance such as pre-eclampsia, inter-pregnancy at high altitude, or maternal respiratory diseases. More significantly, one-third of women are affected by obstructive sleep apnea hypoventilation syndrome (OSAHS) in late pregnancy, and it is also associated with the development of gestational hypertension syndrome. The most critical pathophysiological changes in OSAHS is chronic intermittent hypoxia (3, 4). In several animal models, intrauterine hypoxic (IUH) lead to cardiac efficiency reduction, diastolic and systolic dysfunction, hypertrophic growth, decreased cardiomyocyte proliferation, and delayed cardiomyocyte maturation in the fetal and neonatal heart, thereby increasing the risk of heart disorders in adult offspring (5). The programming effect of chronic hypoxia can mainly explain the mechanism at critical stages of the development of the heart, which leads to cardiac oxidative stress enhancement and cell apoptosis increase in offspring (6-9). The concept of developmental programming makes sense because our physiology is much more malleable and plastic during early life. The intrauterine condition determines and programs the concepts of physiology and metabolism throughout life. Accordingly, the fetal adaptive response to IUH leads to the development of adult cardiovascular diseases (10). Mitochondria are complex organelles playing critical roles in cellular energy-generating and a myriad of cellular signaling events. The structural and functional integrity of mitochondria is critical for the survival of cardiomyocytes, and when compromised, the disruption of mitochondrial homeostasis results in the development of cardiac diseases. Recently, several studies have focused on the role of chronic prenatal hypoxia-inducing mitochondrial dysfunction in mediating offspring heart damage. It has been reported that mitochondrial respiratory function was impaired, and the expression of several mitochondrial molecules altered the perinatal heart exposed to IUH (11). Moreover, IUH leads to decreased mitochondrial complex enzyme activity and the systolic dysfunction of the heart in adult offspring following myocardial ischemia (12). Accordingly, this study aimed to discover the molecular link among prenatal hypoxia, oxidative stress, mitochondrial dysfunction, and heart injury in new offspring (13).
The development and maturation of this high-volume mitochondrial system in the myocardium mainly occur in the perinatal and postnatal development stages, primarily due to the cardiac metabolic shift from using glucose to fatty acids for the adenosine 5'-triphosphate (ATP) generation after birth (14). Emerging evidence suggests mitochondrial quality control (MQC) mechanisms are critical determinants for the maturation of cardiomyocytes (15). Mitochondrial quality control, mainly including mitochondrial biogenesis, mitophagy together with dynamics, a finely tuned regulatory network, orchestrates the quantity and quality of mitochondria and improves mitochondrial function and cardiomyocyte survival under stress conditions (16). Peroxisome proliferators-activate receptors γ (PPARγ) coactivator 1 (PGC-1) play a critical role in driving mitochondrial biogenesis and function in the heart (17). Peroxisome proliferators-activate receptors γ coactivator 1 α/β (-/-) mice hearts exhibits the signatures of the maturational defect and severe abnormality in mitochondrial function and density (18). Furthermore, deleting the mitophagy-mediator Parkin prevents morphological and functional mitochondria maturation in the neonatal stage (19). Mitochondria are highly dynamic organelles, undergoing constant fission and fusion events associated with their function. At the late embryonic period, the cardiac-specific genetic ablation of mitochondrial fusion protein MFN1 and MFN2 in mice display severe mitochondrial dysfunction after birth and develop cardiomyopathy (20). Strikingly, a recent study revealed that chronic hypoxia disrupted mitochondrial dynamics in the fetal guinea pig forebrain (21). Accordingly, MQC may act as a focal point in myocardium injury development in the IUH neonatal offspring. However, little is known about the effects of prenatal hypoxia on the neonatal cardiac MQC system and the effect of MQC dysfunction on neonatal heart health. This study took over this mission to explore the mitochondrial-related mechanism of IUH-induced myocardial damage in new offspring and further explore preventative strategies to reduce the cardiac injury of IUH.
Polyamines (PAs), including putrescine (PU), spermidine (SPD), and spermine (SP), are present in nearly all living organisms and are essential for embryonic and fetal survival, growth, and development (22-24). Studies found that sheep exposed to IUH can improve embryonic dysplasia by supplementing exogenous polyamines (25, 26). Furthermore, the growing body of literature indicates the role of polyamines in scavenging free radicals and protecting DNA, proteins, and lipids from the detrimental damage of oxidative stress. It is also revealed that polyamine supplementation increases life span in model organisms via antioxidant, anti-inflammatory and mitophagy-inducing properties (27-29). Recent studies have documented that supplementation with SPD in dietary is cardioprotective and prolongs lifespan in both mice and humans by stimulating mitophagy and mitochondrial respiration and improving heart function (30, 31). More importantly, we previously reported that maternal hypoxic exposure during the late stages of fetal development resulted in the decreased anabolism and increased catabolism of polyamines in the cardiac tissue of newborn rats. SPD prevented heart injury in seven-day offspring rats exposed to IUH by inhibiting mitochondrial fragmentation (32).
2. Objectives
In this study, IUH is hypothesized to induce the mitochondrial structure and function deficits by increasing oxidative stress and destroying MQC mechanism in the newborn offspring's heart, and maternal SPD treatment in utero is assumed to reduce myocardial injury by decreasing the development programmed of mitochondria. This study may contribute to the development of preventative or therapeutic strategies for IUH offspring to prevent adult cardiovascular diseases.
3. Methods
3.1. Animals
The male and female Wistar rats (3 months old) were purchased from the Department of Laboratory Animal in the Harbin Medical University. All procedures were approved by the Ethics Review Committee of the Harbin Medical University (China), and all experiments were conducted in accordance with the National Institutes of Health guidelines. The rats with a male-to-female ratio of 2: 1 were randomly placed in a cage for mating. Vaginal smears were performed the following day to detect the presence of sperm in vaginal plugs or vaginal smears, which was confirmed as the zero day of pregnancy. The pregnant rats were kept in a room with controlled humidity (60%) and controlled temperature (21°C), and the light-dark cycle was 12: 12 hours.
3.2. Intrauterine Hypoxia Model
From the 15th to the 21st day of pregnancy, the rats in the hypoxia group (n = 10) were put into a closed plexiglass chamber, injected with air and nitrogen, monitored by an oxygen analyzer (Pro OX120; BioSpherix, New York, USA), and inhaled with an oxygen content of 10% for four hours per day. The arterial blood samples were taken from the right femoral artery, the blood gas and pH values were measured to maintain the arterial oxygen partial pressure of 50 - 55 mmHg, the blood oxygen saturation was maintained at 80 - 85%, and specific procedures were performed as previously described (32). The experimental female rats were randomly divided into four groups: control group (control), intrauterine hypoxia group (Hpx), intrauterine hypoxia + spermidine group (Hpx-Spd), and the intrauterine hypoxia + spermidine + inhibitor group (Hpx-Spd-DFMO). Six rats per group were intraperitoneally injected on the 15th - 21st day of pregnancy. The rats were given 0.9% saline (1 mL/kg/d) in the control and Hpx groups, SPD (5 mg/kg/d) in the Hpx-Spd group, and SPD (5 mg/kg/d) and difuoromethy-L-ornithine (DFMO, an inhibitor of the key enzyme of polyamine synthesis ODC) (5 mg/kg/d) in the Hpx-Spd-DFMO group, respectively. After delivery, 1-day-old newborns were sacrificed, and their hearts were extracted for follow-up experimental studies.
3.3. Histological Analysis
The left ventricular tissue of the rats was cut into a thickness of < 1 cm, fixed with 4% paraformaldehyde, dehydrated with alcohol, and embedded in paraffin. The embedded paraffin was cut into 5 mm thick slices, then dried at constant oven temperature of 60°C, dewaxed with xylene, and followed by staining hematoxylin-eosin (HE). The tissue slices were observed to assess the changes in cardiac morphology and structures with an optical microscope (Eclipse E200; Nikon, Tokyo, Japan).
3.4. Immunofluorescence Analysis
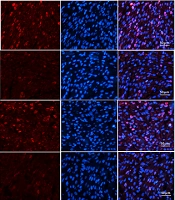
Ki67 immunofluorescence staining was performed as previously described (33). Briefly, the left ventricular tissue of the rats was fixed in 4% formalin, embedded in paraffin; dewaxing, hydration, and antigen repair were first completed. Those tissues were blocked with 0.5% bovine serum albumin for 2 hours and then incubated with Ki67 Rabbit Monoclonal Antibody (1: 100, AF1738, Beyotime, China) at 4°C overnight. After washing with PBS, the tissue was incubated with Alexa fluor labeled Goat anti-rabbit IgG (1: 500, A 0468, Beyotime, China) and counterstained with DAPI for nuclei. The images were viewed and scanned under laser confocal microscopy (OLYMPUS, FV1000, Japan). The software was used to analyze the colocalization of the merged images.
3.5. Quantification of Fibrosis
Cardiac fibrosis was assessed by Masson's trichrome staining. As described above, the cardiac tissue sections from the neonatal rats were dewaxed and hydrated by standard methods and then stained with Masson's trichrome according to the protocols. Two nonadjacent cross-sections were used for each heart. The percentage of fibrotic area in the total left ventricular myocardial area was analyzed using ImageJ software, vl.52 (NIH, Bethesda, MD).
3.6. TdT-Mediated dUTP Nick End Labeling and Apoptotic Cell Measurements
A TdT-mediated dUTP nick end labeling (TUNEL) assay was used to determine the number of apoptotic cells in the one-day-old neonatal rat hearts. The heart tissues were treated following our previously described procedure using a Cell Death Detection Kit (Roche, Germany) according to the manufacturer's instructions. Three slides from each block were evaluated for the percentage of apoptotic cells. Four slide fields were randomly examined with 200 × magnification. In total, 100 cells were counted in each field.
3.7. Measurement of Antioxidant Enzyme Activity
Superoxide dismutase (SOD) and catalase (CAT) activity were measured using commercial kits (SOD: A001-3-1 and CAT: A007-1-1; Jiancheng Bio. Institute, Nanjing, China) with a spectrophotometer (Perkin- Elmer, Norwalk, CT, United States). According to the manufacturer's instructions, the operation was completed, and the protein concentration was measured using the bicinchoninic acid method (Pierce, Rockford, United States) with bovine serum albumin (BSA) as a standard (34).
3.8. Measurement of Adenosine 5'-Triphosphate Content
The ATP content in cardiac tissue was measured using an ATP assay kit (S0026B, Beyotime, Bio. Institute, China). According to the manufacturer's instructions, the lysate was added in proportion according to the tissue weight, homogenized with a glass homogenizer, and then centrifuged at 4°C 12000 g. The supernatant was taken, and the content of ATP in each sample was detected with a luminometer (Nano Drop, Nanodrop2000, Thermo, USA) BCA kit (P0012s, Beyotime, Bio. Institute, China). The bicinchoninic acid method was used to determine the protein concentration and then convert the concentration of ATP into nmol/g protein.
3.9. Transmission Electron Microscopy
The cardiac apical tissue was dissected into approximately 1 mm × 1 mm × 1 mm small pieces and then fixed in glutaraldehyde phosphate buffer at 4°C. After routine dehydration, soaking, embedding, and staining, the ultrathin sections of 50 - 70 nm were made. The ultrastructure of the cardiac tissue was observed under a transmission electron microscope (TEM) and photographed (H600 Hitachi, Tokyo, Japan). Single mitochondria and myofilaments were mapped under the condition of 10000 times magnification with Image J software version 1.80 (National Institutes of Health), and their areas were measured from each heart (35). Meantime, mitochondrial fragments < 1 µm3, which were not divided (usually round), were identified, and the average percentage of mitochondrial fragments in the view field was counted using the Mitochondrial Fragmentation Index (MFI).
3.10. Mitochondrial Isolation
Mitochondria were isolated at 4°C using differential centrifugation with a Mitochondria Isolation Kit (Beyotime Biotechnology, Shanghai, China). Briefly, fresh heart tissue was cut into tissue pieces and centrifuged with 10 volumes of pre-chilled PBS, the supernatant was discarded, the precipitate was digested with trypsin for 20 min, the added isolation buffer A was centrifuged, the supernatant was transferred to another tube, centrifuged again, and the precipitated fraction was the isolated mitochondria. The final cardiac mitochondrial pellet was resuspended in homogenizing buffer, stored on ice, and used for experiments of mitochondrial respiration function within 4h.
3.11. Measurement of Mitochondrial Oxygen Consumption
Mitochondrial oxygen consumption was measured by a Clark-type oxygen electrode (Hansatech Instruments, Norfolk, UK) in mitochondrial respiration buffer. Pyruvate (5 mM) and Malate (5 mM) were used as a substrate for complex I-containing mitochondria at the final concentration of 500 μg protein/mL. ADP-stimulated oxygen consumption (state 3 respiration) was measured in the presence of 200 μM ADP, and ADP-independent oxygen consumption (state 4 respiration) was monitored. The respiratory control ratio (RCR, state 3 divided by state 4) reflects oxygen consumption by phosphorylation (coupling). Procedures continued, as previously described (36).
3.12. Western Blot Analysis
The sample of cardiac tissues was harvested and stored at -80°C. Frozen left ventricular cardiac tissues were homogenized in ice-cold RIPA lysis buffer (Beyotime Inc., Shanghai, China P0013B). The protein concentrations were quantified using a BCA protein assay kit (Beyotime Inc., Shanghai, China P0006C). The samples containing total protein were separated by 10% (w/v) SDS-PAGE and transferred onto a PVDF membrane (Millipore, Bedford, MA, United States). The following antibodies were used: Antibodies for GAPDH (1:2000,10494-1-AP), MFN2 (1:1000,12186-1-AP), SIRT-1 (1:1000,13161-1-AP), NRF-2 (1:600,16396-1-AP), TFAM (1:1000,19998-1-AP), and BAX (1.1000,509599-2-Ig) were purchased from Proteintech (WuHan, China), BCL2 (1: 2000, sc-7382) and DRP1 (1:1000, sc-271583) were purchased from Santa Cruz Biotechnology (Dallas, TX) and PGC-1α (1:1000, ab106814, Abcam, Cambridge, MA, UK). The secondary antibody (horseradish peroxidase-labeled goat anti-rabbit IgG) was from Beyotime Corporation (Shanghai, China). The intensities of the protein bands were quantified using a Fluor Chem Chemiluminescence gel imaging system (Protein Simple, USA). The optical density of the protein bands was analyzed with Image J software version l.52 (NIH, Bethesda, MD).
3.13. Hypoxic Cardiomyocyte Model
Neonatal rat cardiomyocytes (NRMCs) were isolated and cultured by standard methods, as previously described (32). Briefly, the hearts of the three-day neonatal rats were extracted, minced, cultured, and then digested in 0.25% trypsin and 0.02% EDTA (Beyotime Biotechnology, Shanghai, China). After centrifugation, the precipitates were transferred to DMEM supplemented with 10% fetal calf serum (Biolot, Russia) and incubated in humid air containing 5% СО2. Three days after being seeded, the cells were placed in a glass hypoxic chamber (Biospherix OxyCycler C42, Redfield, NY), filled with nitrogen for 8 min to discharge the residual oxygen. These cardiomyocytes were randomly divided into the following groups: (1) Control group (control), cells cultured under normal incubation conditions; (2) hypoxia (Hpx) group, cells placed in the hypoxic chamber for 24 h and then cultured normally for 24 h; (3) Hpx-Spd group, cells placed in the hypoxic chamber and incubated with 10 μmol/L SPD for 24 h; (4) Hpx-Spd-DFMO group, cells placed in a hypoxic chamber and incubated with 10 μmol/L SPD + 2 mmol/L DFMO for 24h.
3.14. Measurement of Reactive Oxygen Species
The ROS generation was measured by the dihydroethidium (DHE) staining assay (Cat No. S0063, Beyotime, China). Briefly, primary cardiomyocytes were incubated with 5 μmol/L DHE at 37°C for 30 min, washed with PBS, and the moved under the microscope to observe the changes in fluorescence intensity. Images were taken with an Olympus FluoView FV1000 fluorescence microscope (Olympus Optical Co., Ltd., Takachiho, Japan) at an excitation wavelength of 535 nm, and the maximum emission wavelength was 610 nm, n > 20 cells per group.
3.15. Determination of Mitochondrial Membrane Potential
The dye tetramethylrhodamine ethyl ester (TMRE) is positively charged and can be selectively located in mitochondria. It is widely used to determine mitochondrial membrane potential (ΔΨm). In short, TMRE staining working solution at a concentration of 200 μmol/L was added to the grouped treated cardiomyocytes, mixed thoroughly, and placed in a cell incubator at 37°C for 20 min. The supernatant was aspirated, and the cells were washed with PBS and moved to an inverted microscope to observe the change in fluorescence intensity. The excitation wavelength was 549 nm, and the emission light was 579 nm. The red fluorescence intensity indicated the change of ΔΨm, n > 20 cells per group.
3.16. Mitochondrial and Lysosomal Localization Experiment
According to the instruction, Mito-Tracker Green (NO.C1048, Beyotime, China) and Lyso-Tracker Red (NO.C1046, Beyotime, China) used the mitochondrial and lysosomal colocalization analysis. NRCMs were loaded with 200 nM MitoTracker Green FM and 50 nM LysoTracker Red in HBSS for 30 min before experiments, cells were washed with PBS, and then cell images were obtained using an Olympus FluoView FV1000 fluorescence microscope. The 488 nm laser line was used to excite MitoTracker Green fluorescence, measured between 505 and 515 nm. For LysoTracker Red, the 577 nm laser line was used with a measurement of 590 nm. Red and green pixel intensity overlay was determined using quantification software on the Nikon eclipse Microscope, n > 20 cells per group.
3.17. Statistical Analysis
All data from the experimental groups were compared using one-way ANOVA followed by Bonferroni post hoc test with GraphPad Prism software version 8 (GraphPad Software Inc., La, Jolla.CA) and SPSS software version 17.1 (SPSS, Chicago, IL, United States). Data were expressed as mean ± SEM, and the significance level was set to be P < 0.05.
4. Results
4.1. Offspring and Heart Characteristics
Body weight (BW) and heart weight (HW) were measured, and the HW to BW ratio (HW/BW) was calculated (Figure 1). The results showed that the newborn rats' BW and HW decreased, and their HW/BW increased due to intrauterine hypoxia. Compared to the IUH group, the BW and HW of neonatal rats in the SPD group increased (P < 0.05), and the HW/BW decreased (P < 0.05); Compared to the SPD group, both BW and HW of the DFMO treatment group decreased (P < 0.05), and HW/BW increased significantly (P < 0.05).
, heart weight (HW) and (HW/BW) ratio of neonatal rats (data are shown as the mean ± SEM; n = 12 per group; * P < 0.05 versus Control, # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the HpxSpd group).")
Effect of prenatal hypoxia on body weight (BW), heart weight (HW) and (HW/BW) ratio of neonatal rats (data are shown as the mean ± SEM; n = 12 per group; * P < 0.05 versus Control, # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the HpxSpd group).
4.2. Effects of SPD on Myocardial Morphological Structure, Cell Proliferation, and Fibrosis in Newborn Offspring Exposed to IUH
The cardiac HE staining results showed that the hearts of one-day offspring exposed to IUH displayed swelling and loosely arranged myocardial fibers. However, the IUH hearts treated with SPD maintained an acceptable myocardial tissue structure (Figure 2A). Next, we evaluated the number of binuclear cardiomyocytes with the HE-stained tissue slice; the number of binuclear cardiomyocytes was larger in the IUH group than in the control group (P < 0.05). Compared to the IUH group, the proportion of binuclear cardiomyocytes in the SPD treatment group decreased significantly (P < 0.05); DFMO attenuated the effect of SPD (P < 0.05) (Figure 2C). We further detected the expression of Ki67 (a marker of cell proliferating) in rat myocardium using the immunofluorescence method. The higher the expression of Ki67, the stronger the pink fluorescence by merging red and blue (Figure 2E). The results showed that compared to the control group, the expression of Ki67 in the IUH group decreased significantly (P < 0.05), the Ki67 expression significantly increased following the SPD administration (P < 0.05), and the effect of SPD was abolished by DFMO (P < 0.05) (Figure 2F). These results indicate that SPD can inhibit the premature withdrawal of cardiomyocytes from the cell cycle induced by IUH and promote the proliferation of cardiomyocytes in the new offspring exposed to IUH. Then we used Masson staining to detect the changes in myocardial collagen content and assessed myocardial fibrosis by collagen area measurement (Figure 2B and D). We found that myocardial collagen deposition in the hearts of the newborn rats exposed to IUH increased (P < 0.05), the level of which was higher compared to the control group. On the contrary, the area of myocardial fibrosis following the SPD treatment significantly reduced (P < 0.05). However, compared to the SPD treatment group, the fibrosis area significantly increased in the Hpx-Spd-DFMO group (P < 0.05).
; D, Evaluation of interstitial fibrotic areas in ventricle sections in each group. (n = 8); E, Immunofluorescence for Ki67 in each group; F, Quantification of the percentage of Ki67 positive cardiomyocytes (n = 8) (data are shown as the mean ± SEM; * P < 0.05 versus control; # P < 0.05 versus the Hpx group; and Δ P < 0.05 versus the Hpx-Spd group).")
The effects of SPD on myocardial morphology structure, cell proliferations and fibrosis in one-day IUH offspring rats. A, Morphological structure changes of myocardial tissue with hematoxylin-eosin staining; B, Representative Masson's trichrome staining in ventricle sections; C, Evaluation of the percentage of binucleated cardiomyocytes (n = 8); D, Evaluation of interstitial fibrotic areas in ventricle sections in each group. (n = 8); E, Immunofluorescence for Ki67 in each group; F, Quantification of the percentage of Ki67 positive cardiomyocytes (n = 8) (data are shown as the mean ± SEM; * P < 0.05 versus control; # P < 0.05 versus the Hpx group; and Δ P < 0.05 versus the Hpx-Spd group).
4.3. Effects of Spermidine on Myocardial Mitochondrial Structure, Respiratory Function, and Adenosine 5'-Triphosphate Content in Newborn Offspring Exposed to Intrauterine Hypoxia
The ultrastructural structure changes of cardiac tissue and mitochondrial characteristic were analyzed by TEM (Figure 3A). Image J software was used to quantify the mitochondrial content percentage (mitochondrial area in the whole cell area) and mitochondrial area (Figure 3B and C). The results showed that in the control group, the myocardial myofilaments were orderly arranged, the sarcomere structure was clear, the mitochondria were compact, the matrix was denser, and the mitochondrial cristae were orderly arranged. However, mitochondria swelled, the loosening matrix and decreased density were observed in some cardiomyocytes of the IUH group. Compared to the control group, the proportion of mitochondria in cardiomyocytes and the area of mitochondria both reduced (P < 0.05). However, in the rat hearts of the SPD treatment, the myocardial myofilaments were neat, the sarcomere structure was clear, the mitochondrial matrix was compact, and the mitochondrial swelling decreased. Compared to the IUH group, the proportion of mitochondria in cardiomyocytes increased (P < 0.05), and the area of mitochondria increased (P < 0.05). DFMO inhibited these SPD-induced effects (P < 0.05).
; B and C, Quantification of the area of cells occupied by mitochondrial (and the quantitative mitochondrial area (n = 10); D - F, Mitochondrial respiratory function was evaluated based on mitochondrial state 3 (D) and state 4 (E) oxygen consumption and the respiratory control rate (RCR) (F); G, Determination of ATP content in cardiac tissue by colorimetry (n = 8) (data are shown as the mean ± SEM; * P < 0.05 versus control, # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the Hpx-Spd group).")
The effects of SPD on cardiac mitochondrial structure, respiratory function and ATP content in neonatal offspring exposed to IUH. A, Ultrastructure changes of the ventricular tissue by transmission electron microscopic observation (magnification, × 20,000); B and C, Quantification of the area of cells occupied by mitochondrial (and the quantitative mitochondrial area (n = 10); D - F, Mitochondrial respiratory function was evaluated based on mitochondrial state 3 (D) and state 4 (E) oxygen consumption and the respiratory control rate (RCR) (F); G, Determination of ATP content in cardiac tissue by colorimetry (n = 8) (data are shown as the mean ± SEM; * P < 0.05 versus control, # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the Hpx-Spd group).
We used pyruvate/malate as the substrate to evaluate the mitochondrial respiratory function, including states 3 and 4 respiratory rates and RCR (Figure 3D - F). We noticed that compared to the control group, the respiratory rate in the states 3 and 4 and the RCR of the IUH group were all significantly lower (P < 0.05). Interestingly, the RCR of states 3 and 4 recovered following the SPD treatment (P < 0.05). In contrast, these SPD effects were significantly inhibited in the DFMO treatment group (P < 0.05). Similarly, compared to the control group, the myocardial ATP content in the IUH group significantly reduced (P < 0.05). Compared to the IUH group, the ATP content remarkably increased in the SPD-treated group (P < 0.05), and DFMO attenuated the effects of SPD (P < 0.05) (Figure 3G). These findings suggest that SPD can protect myocardial mitochondrial structure and function damage and prevent the decline of the ATP levels in neonatal offspring rats exposed to IUH.
4.4. Effects of Spermidine on Myocardial Antioxidative Activity and Apoptosis in Newborn Offspring Exposed to Intrauterine Hypoxia
We measured the activities of antioxidant enzymes SOD and CAT in the myocardium by colorimetry (Figure 4A and B). It was found that the activities of SOD and CAT in the IUH group were significantly lower than those in the control group (P < 0.05); Compared to the IUH group, the activities of SOD and CAT in the SPD group increased significantly (P < 0.05), and DFMO abolished the effect of SPD (P < 0.05). Subsequently, we evaluated cell apoptosis by detecting TUNEL-positive nuclei (Figure 4C and D) and the expression of pro-apoptosis protein BAX and anti-apoptosis BCl-2 (Figure 4E and F). We noticed that the number of TUNEL-positive cardiomyocytes and the BAX/BCL2 protein expression ratio were substantially higher in the IUH group than those in the control group (P < 0.05). In contrast, the ratio of TUNEL-positive cells and BAX/BCL2 protein expression ratio were lower in the myocardium of SPD-treated rats than in the IUH group (P < 0.05). DFMO eliminated the effect of SPD (P < 0.05). To sum up, SPD given in utero can reverse the decrease of antioxidant enzyme activities and the increase of cell apoptosis in offspring hearts induced by IUH.
; C, Brown-stained nuclei indicate TUNEL-positive cells. d. The percentage of TUNEL-positive nuclei in different groups (n = 10); E, BAX and BCL2 protein expression detected by western blotting; F, Quantification of the BAX and BCL2 protein level ration. (n = 6) (data are shown as the mean ± SEM; * P < 0.05 versus control, # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the Hpx-Spd group).")
The effects of spermidine on myocardial oxidative stress and apoptosis in offspring exposed IUH. A and B, Determination of superoxide dismutase and CAT activity in rat myocardium by colorimetry (n = 12); C, Brown-stained nuclei indicate TUNEL-positive cells. d. The percentage of TUNEL-positive nuclei in different groups (n = 10); E, BAX and BCL2 protein expression detected by western blotting; F, Quantification of the BAX and BCL2 protein level ration. (n = 6) (data are shown as the mean ± SEM; * P < 0.05 versus control, # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the Hpx-Spd group).
4.5. Effects of Spermidine on Production of Reactive Oxygen Species, Mitochondrial Membrane Potential, and Mitophagy in Cardiomyocytes Exposed to Hypoxia
Mitochondrial dysfunction is mainly manifested as an increase in ROS production, a decrease in the ATP synthesis, and the accumulation of dysfunctional mitochondria. Mitochondrial membrane potential (ΔΨm) is the proton electrochemical gradient (H+ concentration gradient and transmembrane potential difference) across the mitochondrial inner membrane, which is formed during the electron transfer of mitochondrial oxidative respiratory chain, in which a small number of ROSs are produced. Accordingly, the ROS level and the ΔΨm changes are the main indicators of the mitochondrial function. Excessive ROS production results in the accumulation of dysfunctional mitochondria; the damaged mitochondrial are mainly removed by mitophagy. To investigate whether exogenous SPD could affect the integrity of mitochondrial function in cardiomyocytes under hypoxia conditions, we first examined the ROS levels using a DHE fluorescence probe (Figure 5A). We observed that the ROS production increased after cardiomyocytes exposed to hypoxia for 24 h (P < 0.05), and that SPD inhibited the hypoxia-induced ROS production (P < 0.05); however, DFMO abolished the decrease of ROS production mediated by SPD when DFMO were supplied to cardiomyocytes treated by SPD (P < 0.05) (Figure 5C). Furthermore, ΔΨm was detected using TMRE fluorescent probe (Figure 5B). The stronger the red fluorescence intensity, the higher the mitochondrial potential (P < 0.05). We noticed that the intensity of red fluorescence was significantly lower in the IUH group than in the control group (P < 0.05). Compared to the IUH group, the red fluorescence intensity was higher in the SPD treatment (P < 0.05), and DFMO abolished the effects of SPD once more (P < 0.05) (Figure 5D). Then we estimated the colocalization of the mitochondria (labeled with Mito-Tracker green) with the lysosomes (labeled by Lyso-Tracker red), which allowed us to measure the autophagic degradation of mitochondria (Figure 5E). As indicated in Figure 5F, compared to the control, hypoxia decreased the colocalization of Mito-Tracker Green and Lyso-Tracker Red in cardiomyocytes (P < 0.05). However, compared to the hypoxia group, the percentage of colocalized mitochondria and lysosomes was higher in the SPD treatment group (P < 0.05), and DFMO abolished the effect of SPD (P < 0.05). These results suggest that hypoxia increases the ROS production and decreases ΔΨm and mitophagy; SPD can alleviate hypoxia-induced mitochondrial damage by stimulating mitophagy and inhibiting oxidative stress.
 was detected by TMER fluorescent probe; C, Statistical quantification of the average fluorescence intensity of DHE RED; D, Statistical quantification of the average fluorescence intensity of TMRE RED; E, Representative images of lysosomes labeled with LysoTracker Red (red) and mitochondria with Mito-Tracker Green (green). Merging of both signals is counted as a mitophagy event; F, Histogram showing co-localization of Lyso-Tracker Red with Mito-Tracker Green in cardiomyocytes, as a sign of mitophagy (the presented picture is a representative picture from six independent experiments; Data are shown as the mean ± SEM; * P < 0.05 versus control; # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the Hpx-Spd group).")
The effects of spermidine on oxidative stress, apoptosis and mitophagy in cardiomyocytes exposed to hypoxia. A, Reactive oxygen species production was detection in cardiomyocyte by DHE fluorescent probe; B, Mitochondrial transmembrane potential (ΔΨm) was detected by TMER fluorescent probe; C, Statistical quantification of the average fluorescence intensity of DHE RED; D, Statistical quantification of the average fluorescence intensity of TMRE RED; E, Representative images of lysosomes labeled with LysoTracker Red (red) and mitochondria with Mito-Tracker Green (green). Merging of both signals is counted as a mitophagy event; F, Histogram showing co-localization of Lyso-Tracker Red with Mito-Tracker Green in cardiomyocytes, as a sign of mitophagy (the presented picture is a representative picture from six independent experiments; Data are shown as the mean ± SEM; * P < 0.05 versus control; # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the Hpx-Spd group).
4.6. Effects of SPD on myocardial MQC system in newborn offspring exposed to IUH
Mitochondrial quality control (MQC), including the processes of mitochondrial autophagy, mitochondrial biosynthesis, and mitochondrial fusion and fission, is critical for maintaining the normal morphology, quantity, and function of mitochondria and ensuring the normal function and metabolism of cells. Mitophagy regulates MQC and mediates extensive autophagic degradation of mitochondria in response to patho-and physiological alteration. We first evaluated mitophagy by observing the formation of mitophagosome with TEM (Figure 6A - C). According to the results, some abnormal structures or fragmentation mitochondria were enclosed within a double membrane autophagic membrane, which were mitophagosomes and indicated that mitophagy was active in the control group. However, the number of mitophagosomes highly decreased in the myocardium of the IUH group. Interestingly, the myocardium of the SDP group showed almost identical mitophagy phenotypes to that of the control group, while DFMO abolished the effects induced by SPD. Mitochondria are highly dynamic organelles, undergoing constant fission and fusion events associated with their function. We next examined mitochondrial fragmentation using TEM (Figure 6A). We found that the numbers of small and fragment mitochondria significantly increased by IUH, compared to the control (P < 0.05), indicating that mitochondrial fission was broadly raised. This alteration was restrained by the SPD treatment, and DFMO also abolished the role of SPD once more (Figure 6D). Furthermore, DRP1 and MFN2 (regulating proteins of mitochondrial dynamics) were detected (Figure 6E - G). MFN2 significantly decreased, whereas DRP1 increased and observed in the IUH group compared to the control group (P < 0.05). Whereas DRP1 was down-regulated and MFN2 was upregulated with the treatment of exogenous SPD (P < 0.05), DFMO abolished the effects induced by SPD. Mitochondrial biosynthesis coordinates with mitophagy to remove damaged or excess mitochondria, generate new mitochondria and maintain the homeostasis of MQC under stress conditions. Finally, we evaluated the expression of mitochondrial biosynthesis-related proteins, including SIRT-1, PGC-1 α, NRF-2, and TFAM, by the immunoblot analysis (Figure 6H - L). Compared to the control group, the expression of SIRT-1, PGC-1 α, NRF-2, and TFAM decreased significantly in the myocardium of the IHU group (P < 0.05). Compared to the IUH group, the protein expressions of SIRT-1, PGC-1 α, NRF-2, and TFAM in the SPD group significantly increased (P < 0.05). DFMO abolished the effect of SPD (P < 0.05). To sum up, these results indicate that IUH leads to MQC dysfunction in the myocardial of one-day-old offspring, and that the SPD treatment significantly inhibits the imbalance of the MQC system in neonatal offspring exposed to IUH.
; C, Quantitative measurement of cardiomyocyte mitophagosome; D, Quantitative measurement of cardiomyocyte mitochondrial fragmentation index; E, The expression of the proteins DRP1 and MFN2 in each group were assessed by western blotting; F, G, Quantification of DRP1and MFN2 protein levels (n = 4); H, The expression of the protein STIR-1, PGC-1α, NRF-2 and TFAM were assessed by western blotting; I - L, Quantification of STIR-1, PGC-1α, NRF-2 and TFAM protein levels (n = 4) (data are shown as the mean ± SEM; * P < 0.05 versus control, # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the Hpx-Spd group).")
The effects of SPD on cardiac mitophagy, mitochondrial biogenesis and mitochondrial fusion and fission in newborn offspring exposed to IUH. A, B, Representative transmission electron microscopy images showing structural changes of mitochondria in the myocardia (magnification, 10,000 X, magnification, 60,000 X); C, Quantitative measurement of cardiomyocyte mitophagosome; D, Quantitative measurement of cardiomyocyte mitochondrial fragmentation index; E, The expression of the proteins DRP1 and MFN2 in each group were assessed by western blotting; F, G, Quantification of DRP1and MFN2 protein levels (n = 4); H, The expression of the protein STIR-1, PGC-1α, NRF-2 and TFAM were assessed by western blotting; I - L, Quantification of STIR-1, PGC-1α, NRF-2 and TFAM protein levels (n = 4) (data are shown as the mean ± SEM; * P < 0.05 versus control, # P < 0.05 versus the Hpx group, and Δ P < 0.05 versus the Hpx-Spd group).
5. Discussion
Heart disease is a major health challenge worldwide. It has been demonstrated that chronic fetal hypoxia can trigger a fetal origin of cardiac dysfunction and program an increased risk of heart disease in the adult offspring. The heart has a high metabolic rate, and its reliance on mitochondrial function for energy production is critical for the normal cardiac function. However, the effect of IUH on the cardiac mitochondrial function and its related mechanism and the focus of cardio-protection preventive strategy on mitochondria in the newborn offspring are still unknown. In the present study, we found that one-day offspring rats exposed to IUH exhibit significant abnormalities in growth and development, cardiomyocytes maturation, especially in the myocardial mitochondrial structure and function, and exogenous SPD supplementary in utero can relieve this IUH-induced change by inhibiting myocardial oxidative stress and reducing apoptosis. Importantly, we noticed that prenatal SPD treatment can significantly reverse the abnormalities of mitophagy and biogenesis and mitochondrial fragmentation in the myocardium of newborn offspring rats induced by IUH. To the best of our knowledge, this research was the first report on the protective effect of SPD on IUH-induced newborn offspring’s heart damage by regulating the MQC mechanism and then improving mitochondrial function.
Sufficient evidence suggests that intrauterine growth restriction interferes with the developmental trajectory of fetal systems, which can have a negative impact on heart function later in life (5, 7, 37). In our study, IUH caused growth retardation and heart damage in neonatal offspring rats, as the one-day newborn rats exposed to IUH exhibited a significant decrease in BW, and HW and an increase in the HW/BW ratio. Moreover, the arrangement of myocardial fibers was disordered, myofilaments were broken, inflammatory cell infiltration was noticed among myofilaments, and myocardial collagen deposition was increased. These findings are consistent with findings reported by Ducsay et al. (2) and Thompson et al. (7). In several animal models, fetal hearts exposed to chronic hypoxia exhibit reduced cardiomyocyte maturation and proliferation and decreased cardiomyocyte endowment (38, 39). Similarly, our research also showed that the proportion of binucleate cardiomyocytes increased and the expression of Ki67 decreased in the IUH group. The time at which cardiomyocytes become bi/multi-nucleated coincides with the time when mammals lose their regenerative potential (40). Fetal heart maturation is important for offspring’s heart health. Accordingly, IUH makes cardiomyocytes withdraw from the cell cycle ahead of time, and cardiomyocyte maturation disorder leads to the impaired plasticity of postnatal heart growth.
Needless to note that mitochondria are the main cellular organelles involved in the production of ROS; dysfunctional mitochondria are not only produced in response to decreased ATP production but also to increased oxidative stress and apoptosis (41). In this study, IUH significantly decreased the activity of antioxidative enzyme SOD and CAT, decreased mitochondrial respiratory function and the ATP levels, increased the expression of BAX/BCL2, and impaired the mitochondrial structure in the myocardium of one-day newborn offspring rats. These findings are supported by other researchers (11), who documented that maternal chronic hypoxia exposure during pregnancy could decrease the mitochondrial complex IV activity and induce apoptosis in the hearts of near-term fetal rats. We argued that mitochondrial compromise might underpin the oxidative stress and energy failure during fetal development culminated in perinatal heart injury. This can be the main reason for increased cardiovascular risk in offspring of pregnancies complicated by chronic fetal hypoxia. Accordingly, it is implementable to take mitochondrial as the target of therapeutic intervention to protect cardiac function in the offspring. Aljunaidy et al. reported that maternal treatment with mitochondrial antioxidant MitoQ prevented placental oxidative stress, rescued fetal growth, and improved cardiac function in offspring exposed to IUH (42).
Spermidine is a polyamine with a variety of biological functions. As we mentioned, SPD can inhibit the H2O2-induced ROS accumulation and prevent the decrease in mitochondrial membrane potential (MMP) and ATP levels in cultured neonatal rats’ cardiomyocytes and H9C2 cells. Moreover, SPD can promote mitochondrial biogenesis and improve mitochondrial function in the aging heart (43). Polyamines spermine and spermidine can affect the dynamic mitochondrial proteome, thereby preventing age-related changes in cardiac functions and cardiac aging (36). Other studies have also demonstrated that SPD supplementary delay cardiac aging by enhancing mitophagy and promoting mitochondrial respiration in mice (44). ODC is a key enzyme of polyamine synthesis, and it is reported that the in vivo knockdown of ODC or the application of DFMO, an irreversible inhibitor of ODC results in disrupted conceptus development and fetal growth retardation due to the insufficient availability of polyamines (45, 46). Our present study indicates that treatment with 5 mg/kg/d of SPD in rats exposed to IUH prevents growth retardation and the alteration of cardiac morphological structure and maturation in newborn offspring. Moreover, IUH-induced cardiac oxidative stress enhancement, apoptosis increases, and mitochondrial respiratory decline in neonatal offspring were also prevented by SPD, while DFMO given to the mother abolished the SPD-mediated protection for the neonatal heart. It is well-documented that polyamines involve in placental angiogenesis and embryogenesis as well as embryonic and fetal growth and development (47). Zhu et al. reported that polyamine levels were lower in the umbilical vein plasma from intrauterine growth restriction (IUGR) porcine fetuses, and supplementary SPD to the mother may prevent IUGR (48). Spermidine is a compound with reasonable safety and lower toxicity (49). The increasing numbers of preclinical and clinical studies have indicated that dietary SPD has health effects such as antiaging, cardiovascular protection, neuromodulation, and anti-inflammatory functions, such as intraperitoneal injection of SPD (50 mg/kg) in mice protects against collagen-induced arthritis by inhibiting the polarization of M1 macrophages in synovial tissue (50). Supplementation with 10 mg/kg/d of SPD reversed age-related cardiac deterioration in rats (36). More importantly, the mother was an intervention with 5 mg/kg/d of SPD during late pregnancy to improve IUH-induced cognitive and neural function decline (51) and heart oxidative stress damage in rat offspring (32). Accordingly, we used the same dose of SPD (5 mg/kg/d) for the pregnant rat exposed to IUH as in our previous study. We suspected that maternal SPD supplementation during hypoxic pregnancy might increase SPD levels in fetal circulation, which might have been related to protection against oxidative stress result in the placenta, which will protect the mitochondria in the developing heart from the programming by IUH, the action of SPD may be achieved by its powerful antioxidative, anti-apoptosis, and proliferation-promoting mechanism.
Mitochondrial quality control mechanisms are essential for maintaining the normal morphology, number, and function of mitochondria and ensuring cells' normal function and metabolism (16). Recent evidence has revealed that MQC mechanisms play a decisive role in perinatal myocardial mitochondrial maturation and heart development (14). Gong et al. first reported that the immature fetal-like mitochondria are directly degraded by Parkin-mediated mitophagy during the early postnatal period (19). In the meantime, mitochondrial biogenesis increases dramatically (52), and the mitochondrial fusion and fission factors are transcriptionally upregulated in the heart after birth (20, 21). However, much less is known about the effect of IUH on MCQ in the myocardium of neonatal offspring. In this study, we first observed that the number of mitophagosome decreased significantly in the myocardium of the IUH group, indicating that IUH impaired the myocardial mitophagy of newborn offspring rats. Next, we observed a significantly decreased expression of PGC-1α (a key regulator of mitochondrial biogenesis) and the expression of its upstream and downstream signaling molecules, including SIRT-1, TFAM, and NRF-2 in the myocardium of the IUH group. Moreover, we found that the IUH exposure made smaller or fragmented mitochondria and the expression of DRP1 increase; however, the expression of MFN2 decreased in the myocardium of newborn offspring rat, suggesting that mitochondrial fission was upregulated, and fusion was down-regulated. These findings reflected the cause-and-effect relationship between the imbalance of the MQC mechanism and IUH-induced myocardium damage in newborn offspring. These complex MQC processes function in concert with one another to eliminate dysfunctional mitochondria in a specific and targeted manner and coordinate the biogenesis of new organelles, which is essential for cardiovascular homeostasis. Many intracellular and extracellular signals can regulate the MQC events, including oxidative stress, potential membrane collapse, apoptosis, and others. Here, the imbalance of mitochondrial MQC may be attributed to enhanced oxidative stress and increased apoptosis in the heart of offspring rats exposed to IUH. Accordingly, it is essential to maintain a suitable MQC mechanism to ensure a healthy mitochondrial network in the heart of newborn offspring exposed to IUH.
Given that placental SPD levels are strongly associated with fetal growth restriction (53), SPD can regulate MQC in a different animal model. For example, SPD is a potent and specific inducer of autophagy for expanding lifespan, which can protect the aging process of several tissues, including the heart, brain, and skeletal muscle (44, 54, 55). Spermidine can alleviate cardiac aging by improving mitochondrial biogenesis (43). In this study, we applied SPD to maternal mice and found that SPD could normalize the unbalance of myocardium MQC in offspring exposed to IUH. However, maternal treatment with ODC inhibitor DFMO abolished the effects of SPD, indicating that the SPD intervention could prevent the abnormal MQC in the myocardium of one-day offspring rats exposed to IUH. To sum up, the excellent working of MQC in the myocardium of newborn offspring could make well of mitochondrial structure, function, and maturation, thus realizing the transformation from intrauterine glycolysis capacity to postnatal fatty acid oxidation energy supply and meet the increasing heart load after birth. Furthermore, our experiment on cardiomyocytes suggests that SPD can inhibit the increase in hypoxia-induced ROS production, the decrease in ΔΨm, and the impaired autophagic degradation of mitochondria. We deduced that the beneficial effects of SPD could attribute to the increased SPD levels in placental and circulation by applying maternal SPD, thereby protecting offspring myocardial mitochondrial from development programming by IUH.
5.1. Conclusion
The present study highlights that SPD therapy in utero can protect the myocardium of one-day- the offspring's heart from the adverse effects of prenatal hypoxia. These postnatal changes caused by SPD may be achieved by anti-oxidation and anti-apoptosis and by regulating MQC pathways, thereby decreasing the mitochondrial development programmed in the heart exposed to IUH. There are some limitations to this study. We did not perform pharmacokinetic experiments on SPD, and thus, we cannot determine the exact SPD levels in fetal circulation when the mother was administrated with SPD. In our future studies, we will further investigate the effective blood concentration of SPD using the placenta to the fetus. The present findings provide a new framework for future studies to define the roles of SPD in preventing the myocardial mitochondria of IUH neonatal offspring.
