




1. Introduction
Lipoid proteinosis (LP) is a rare disease of multiple organs and is considered to be an autosomal recessive genodermatosis. Deposition of abnormal amounts of hyaline like material due to a mutation on chromosome 1q21 causes clinical manifestations in the skin and viscera (1). Involvement of skin and viscera including upper digestive and respiratory tracts by hyaline material deposits is seen in lipoid proteinosis or “Urbach-Wiethe” disease (1-3). Approximately 300 cases have been described totally so far (3).
Central nervous system (CNS) involvement is seen in 50% - 75% of individuals. The typical imaging finding in lipoid proteinosis is intracranial, fairly dense calcifications in the bilateral medial temporal lobes. Amygdala involvement is pathognomonic and is more prominent with a longer duration of the disease. Commonly involved sites in computed tomography (CT) scan are the amygdala, hippocampus, parahippocampal gyrus, and the striatum (3). Hoarseness, which is present in approximately two-thirds of the patients at birth or early infancy, is caused by hyaline material infiltration in the larynx (4).
2. Case Presentation
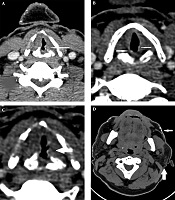
A 43-year-old man was referred to our imaging center in the head and neck hospital with a long history of hoarseness beginning in youth. He also had a history of warty skin lesions. The patient was referred to the hospital for shortness of breath and he did not have any workup previously. On neck CT scan, findings were bilateral true vocal cords (TVCs) mucosal irregularity with hyperdense depositions (Figure 1A-C). Right glottic lesion was visible at laryngoscopy. Biopsy of right TVC showed submucosal deposition of periodic acid-Schiff (PAS)-positive amorphous hyaline material without inflammation, acanthosis, hyperkeratosis, and elongated rete ridges. Other head and neck CT scan findings were bilateral parotids fat replacement and cutaneous punctuate hyperdense depositions (Figure 1D). On brain CT scan, bilateral medial temporal amygdala parallel bean shape calcification (pathognomonic sign) was present (Figure 2A and B). Bilateral striatal (caudate and putamen) hypoattenuation (Figure 2C and D) were also seen. No history of any seizures or focal neurologic deficit was detected. He had multiple warty papules on the dorsal surface of his hands (Figure 3). His sister also has a history of lipoid proteinosis.
 mucosal irregularity with hyperdense deposition (arrows); D, Bilateral parotid fat replacement (wide arrow) and cutaneous punctate hyperdense depositions (narrow arrow).")
A-C, Bilateral true vocal cords (TVC) mucosal irregularity with hyperdense deposition (arrows); D, Bilateral parotid fat replacement (wide arrow) and cutaneous punctate hyperdense depositions (narrow arrow).
; C and D, Bilateral striatal (caudate and putamen) hypo-attenuation (arrows)")
A and B, Bilateral temporal amygdala parallel bean shape calcification (arrows); C and D, Bilateral striatal (caudate and putamen) hypo-attenuation (arrows)
")
Hyperkeratotic yellowish papules on dorsum of the hands (arrows)
3. Discussion
Genetic mutation of extracellular matrix protein 1 (ECM1) encoding gene located on chromosome 1q21 is responsible for a rare autosomal recessive disease called LP (1).
Genotype-phenotype correlation has not been shown for neurologic features (1). This disease shows no race, sex or age tendency (5). Distribution of the disease is worldwide. However, the frequency seems to be higher in countries with higher consanguinity rate such as Germany, India and the Middle East (6, 7).
The disease may present in any part of the body and the clinical manifestations of LP may vary significantly between affected patients. Diffuse infiltration of hyaline material deposits in the mucosal membrane of vocal cords results in first clinical manifestation of LP as a weak cry at birth or progressive hoarseness within early years of childhood as our case (4, 8).
In case of CNS involvement, hippocampal capillaries are common sites of infiltration with wall thickening and subsequent perivascular calcium deposition. Gliotic tissue surrounding amorphous calcification is also seen in microscopy (3). Neurologic manifestations range from migraine, seizure disorder, and depression to anxiety disorder (3).
The typical and pathognomonic imaging findings in LP are seen in bilateral medial temporal lobes and amygdala. Curvilinear or comma-shaped hyperdense symmetrical calcifications are depicted in amygdala and are more notable in longer disease similar to our case (3, 9).
In CT scan, common sites of involvement are amygdala, striatum, hippocampus, and parahippocampal gyrus. On magnetic resonance imaging (MRI), signal intensity of the lesions are low in T1 and T2 and are well brought out in T2* gradient echo (GRE) images. In the absence of brain calcifications, the CT or MRI features of these patients are unremarkable (3, 9). Head and neck imaging manifestation of the disease has not been reported yet. Our case had bilateral TVCs mucosal irregularity with hyperdense depositions and bilateral parotids fat replacement and punctuate hyperdense depositions in the skin.
Recurrent vesicles, pustules, bullae, and hemorrhagic crusts in variable sizes may be seen on the skin, oral and laryngeal cavities, mostly in the face and distal extremities. Another pathognomonic finding, which is present in half of the patients, is moniliform blepharosis, beaded papules lying in a row in the margin of eyelids (8, 10).
In conclusion, imaging findings of LP in the head and neck has not been reported yet and the radiologist should be aware of LP as the differential diagnosis of long-term voice changes and hoarseness with hyperdense depositions in TVC on imaging, and typical imaging findings, especially in the brain could be very helpful (8). It is also important that genetic counseling about risks of having another affected offspring be offered to parents of affected children (10).
