




1. Introduction
Monoclonal antibodies stepped into the limelight in the early 1980s. In 1986, the monoclonal antibody named Orthoclone OKT3 was approved for the prevention of graft rejection in patients who had received kidney transplantation (1). Since then, numerous monoclonal antibody-based products, such as therapeutic monoclonal antibodies, antibody-drug conjugates, immunotoxins, bispecific T-cell engagers (BiTEs®), etc. have been developed and investigated in preclinical and clinical studies (with more than 450, 290, and 88 monoclonal antibodies in phase I, II, and III, respectively) (2, 3). So far, around 100 monoclonal antibodies have been approved by the US Food and Drug Administration (FDA) for various cancer and non-cancer indications (such as severe asthma, homozygous familial hypercholesterolemia, Alzheimer’s disease, Ebola virus infection, etc.) (4-7).
Various techniques have been developed and employed for the isolation of monoclonal antibodies. Such techniques include hybridoma (which entail the fusion of B cells with myeloma cells to establish immortalized cells capable of producing antibodies), phage display (a technique that uses bacteriophages for the expression and screening of antibodies), and ribosome display (which consists of in vitro protein evolution to select a high-affinity antibody for a given antigen target) (8-10). Moreover, a high-affinity monoclonal antibody is usually isolated from a large population of antibodies, which are eliminated in a screening process as an attempt to achieve an antibody with a strong binding capacity to the desired target antigen. This large population of antibodies is produced from a large gene library which can be a naïve or an immunized one (11). Since most of the immunized gene libraries are produced in animals following animal immunization (usually mouse or camel), the resultant monoclonal antibodies could be of high immunogenicity while administered to human subjects as immunotherapeutics (11-14). This could result in the production of neutralizing antibodies against such monoclonal antibodies by the host immune system, which consequently leads to the rapid elimination of the therapeutics from the patient’s body (11-14). In this regard, monoclonal antibodies are humanized as an attempt to reduce their immunogenicity while maintaining their affinity (11-14). In detail, a conventional humanization process, which is known as complementarity-determining region (CDR) grafting, entails the replacement of the animal-based residues of a given monoclonal antibody with those of its human origin (11-14). For instance, the CDRs of a monoclonal antibody are grafted onto the framework regions of an acceptor antibody with the human origin (11-14). Since the CDRs of the monoclonal antibody are untouched during the course of humanization, they could have no effects on its antigen-affinity (11-14). Such methods lead to the production of monoclonal antibodies, which could be utilized for various therapeutic purposes.
Biopharmaceutical companies are taking rapid steps towards developing and investigating monoclonal antibodies, and it has been anticipated that the speed of approval for monoclonal antibodies in the 2020s will be more than the past decades. In this review, we will take a look at the clinical investigations that led to the approval of certain monoclonal antibodies in the 2020s (Table 1).
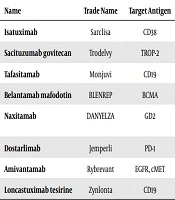
| Name | Trade Name | Target Antigen | Antibody Format | Indication | FDA Approval Date |
|---|---|---|---|---|---|
| Isatuximab | Sarclisa | CD38 | Chimeric IgG1 | Multiple myeloma | 2020 |
| Sacituzumab govitecan | Trodelvy | TROP-2 | Humanized IgG1 ADC | Triple-negative breast cancer | 2020 |
| Tafasitamab | Monjuvi | CD19 | Humanized IgG1 | Diffuse large B-cell lymphoma | 2020 |
| Belantamab mafodotin | BLENREP | BCMA | Humanized IgG1 ADC | Multiple myeloma | 2020 |
| Naxitamab | DANYELZA | GD2 | Humanized IgG1 | High-risk neuroblastoma and refractory osteomedullary disease | 2020 |
| Dostarlimab | Jemperli | PD-1 | Humanized IgG4 | Endometrial cancer | 2021 |
| Amivantamab | Rybrevant | EGFR, cMET | Human bispecific IgG1 | NSCLC with EGFR exon 20 insertion mutations | 2021 |
| Loncastuximab tesirine | Zynlonta | CD19 | Humanized IgG1 ADC | Diffuse large B-cell lymphoma | 2021 |
A summary of Monoclonal Antibodies Approved by the FDA for Cancer Treatment in the 2020s
2. Monoclonal Antibodies Approved in the 2020s
2.1. Isatuximab
Isatuximab (also named SAR650984 or Sarclisa®) is a chimeric IgG1 antibody developed by ImmunoGen and Sanofi-Aventis capable of targeting CD38 (15-20). CD38 is a cell surface antigen with ectoenzymatic activity overexpressed on malignant plasma cells, which are a type of B cells responsible for antibody production and secretion in the body (21). Therefore, this antigen can be a considerable target in the immunotherapy of multiple myeloma. In March 2020, FDA granted approval for the clinical application of isatuximab based on the report from the clinical trial involving 307 patients with relapsed/refractory (R/R) multiple myeloma (MM) (NCT02990338) (15). The data from this trial demonstrated that MM patients treated with isatuximab in combination with pomalidomide/dexamethasone had longer progression-free survival (PFS) than patients only treated with pomalidomide/dexamethasone (15). This trial was a multicenter investigation, which was performed in more than a hundred locations, and the patients enrolled in this trial had been treated with at least two prior treatments, including lenalidomide and a proteasome inhibitor (15). So far, there has not been any severe side effect related to the use of isatuximab (22). However, mild levels of administration-related adverse events, including neutropenia, dyspnea, cough, chills, nausea, and hypertension, have been reported (22). The application of isatuximab as a monotherapy for the treatment of systemic light-chain amyloidosis (AL), which is refractory or recurrent, is currently being investigated in a phase II clinical trial (NCT03499808), which is estimated to be completed in 2025 (22).
2.2. Sacituzumab Govitecan
Sacituzumab govitecan (which is also known as IMMU-132 or TrodelvyTM) is a TROP2-specific ADC consisting of a humanized IgG1 antibody conjugated to an irinotecan metabolite that is recognized as SN-38 (23, 24). In vitro experiments have indicated that the cytotoxicity potential of SN-38 is almost a thousand-fold higher than that of irinotecan. Based on the results of a single-arm clinical trial (clinical trial identifier: NCT01631552), known as IMMU-132-01, with 108 subjects with metastatic triple-negative breast cancer (TNBC), FDA gave sacituzumab govitecan (under the commercial name Trodelvy) accelerated approval in April 2020 (23, 24). This approval is indicated for the treatment of metastatic TNBC individuals who have undergone two or more anti-metastatic treatments (23, 24). In reference to the mentioned trial, sacituzumab govitecan was administered on days 1 and 8 every three weeks as the tumor volume was imaged every 56 days (23, 24). Moreover, the treatment was maintained until the subjects experienced disease progression or became intolerant to sacituzumab govitecan (23, 24). According to the results, the overall response rate (ORR) was between 24.6% and 43.1% (with a median of 33.3%), and the patient response duration was found to be between 4.9 and 10.8 months (with a median of 7.7 months). Moreover, of the patients responsive to sacituzumab govitecan, about half of them remained responsive for more than half a year, and 16.7% retained their response for a year or even more (23, 24). Moreover, the therapeutic efficacy and tolerability of sacituzumab goviteca were also investigated in a single-arm multicenter clinical trial (clinical trial identifier: NCT03547973; IMMU-132-06 or TROPHY-U-01) with 112 locally advanced or metastatic urothelial cancer (mUC) patients who had undergone either PD-1 or PD-L1 inhibitor treatment alongside platinum-based chemotherapy (25). Based on the findings of the mentioned clinical trial, in April 2021, sacituzumab govitecan was granted accelerated approval by the FDA for individuals with locally advanced or mUC with the same condition as of those enrolled in the IMMU-132-06 investigation (25). Of note, sacituzumab goviteca comes with a boxed warning that indicates that severe diarrhea and severe neutropenia might occur following the administration of this ADC (23-25). Moreover, the administration of sacituzumab goviteca has also been intertwined with a series of side effects that include skin rashes, constipation, nausea, anemia, vomiting, and hair loss (23-25).
2.3. Tafasitamab
Tafasitamab (also known as Monjuvi®, tafasitamab-cxix, MOR208, and XmAb5574) is a humanized monoclonal antibody with great targeting ability and specificity towards CD19 developed by MorphoSys AG (26-30). CD19 is a surface antigen that is expressed from the early developmental stages of pro-B-cells until its downregulation at the final differentiation stage of B cells, in which they develop into antibody-secreting plasma cells (26). This antigen has been known as one of the most investigated cancer immunotherapy targets since it has a high expression level in many types of hematological malignancies, including several types of leukemias and lymphomas (26). So far, many CD19-targeting immunotherapeutics have been approved by the FDA for clinical use in patients with R/R hematologic malignancies, such as B-ALL (13, 31-33). This antibody harbors a genetically engineered Fc domain rendering this antibody capable of mediating programmed cellular death and Fc-related cellular mechanisms, such as antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) (26).
In July 2020, FDA approved the medical application of tafasitamab for the treatment of patients with R/R diffuse large B-cell lymphoma (DLBCL). This permission is only granted for medical use in combination with lenalidomide and only in patients who are not considered suitable for receiving autologous stem cell transplantation (34). The FDA approval of tafasitamab was based on the report of a phase II, single-arm, open-label, and multicenter clinical trial (NCT02399085) on 81 participants with R/R DLBCL (34). This trial started in March 2016, and its actual primary completion date was in November 2018 (34). The patients in this study were administered with tafasitamab (12 mg/kg through IV administration) in combination with lenalidomide (25 mg/day through oral administration) followed by tafasitamab as monotherapy (34). Of 80 patients who received tafasitamab plus lenalidomide, 48 (60%) had an objective response, 34 (43%) had a complete response, and 14 (18%) had a partial response (34). Common adverse events reported in this investigation include neutropenia (in 48% of the patients), thrombocytopenia (in 17% of the patients), and febrile neutropenia (in 12% of the patients) (34). Moreover, serious adverse events were also reported in patients enrolled in this trial, including pneumonia (6% of the patients), febrile neutropenia (6% of the patients), pulmonary embolism (4% of the patients), bronchitis (2% of the patients), atrial fibrillation (2% of the patients), and congestive cardiac failure (2% of the patients) (34).
2.4. Belantamab Mafodotin
Belantamab mafodotin (also known as GSK2857916, belantamab mafodotinblmf, and Blenrep®) is a humanized monoclonal antibody conjugated to the cytotoxic agent maleimidocaproyl monomethyl auristatin F (MMAF) developed by GlaxoSmithKline (35-39). MMAF is an antimitotic anticancer agent capable of inhibiting cell division by interfering with the polymerization process of tubulin (40, 41). The antibody of this ADC has a significant affinity towards the B cell maturation antigen (BCMA). BCMA is a cell surface antigen that is overexpressed in malignant plasma cells (42). This antigen has been investigated in many kinds of targeted cancer therapies. CAR T cells redirected against BCMA have received FDA approval for a medical application for the treatment of patients with MM (33). These outcomes highlight the significant potential of BCMA for targeted cancer therapy. In this regard, in August 2020, belantamab mafodotin also received accelerated FDA approval as monotherapy for the treatment of adult patients with MM certain criteria. These criteria include disease development observed on the previous treatment approach, significant response failure in at least four previous therapies, and the refractory status of the disease to at least one proteasome inhibitor, a CD38-redirected monoclonal antibody, and one immunomodulatory agent. The FDA approval of belantamab mafodotin was based on a report from a two-arm, randomized, open-label, Phase II clinical trial (which started in June 2018 and is still ongoing) done at more than 50 multiple myeloma specialty centers in eight different countries.
Patients received IV infusion of belantamab mafodotin (2.5 mg/kg or 3.4 mg/kg) on time every 3 weeks until disease progression or unacceptable toxicity was documented (35). As of June 21, 2019, 31% of the patients (30 patients) in the 2.5 mg/kg cohort and 34% of the patients (33) in the 3.4 mg/kg cohort achieved an overall response (35). The most common adverse events in the safety population include keratopathy (in 27% and 21% of the patients in the 2·5 mg/kg cohort and 3.4 mg/kg cohort, respectively) and thrombocytopenia (in 20% and 33% of the patients in the 2.5 mg/kg cohort and 3.4 mg/kg cohort, respectively), and anemia (in 20% and 25% of the patients in the 2·5 mg/kg cohort and 3.4 mg/kg cohort, respectively) (35). It is worth mentioning that 40% and 47% of the patients in the 2.5 mg/kg cohort and 3.4 mg/kg cohort, respectively, reported having serious adverse events (35). Overall, two treatment-related deaths were also reported, with one of them from the 2.5 mg/kg cohort and the other one from the 3.4 mg/kg cohort (35).
2.5. Naxitamab
Naxitamab is a humanized GD2-specific monoclonal antibody (also known as naxitamab-gqgk or hu3F8), which in November 2020, was approved for the treatment of patients (≥ 1-year-old) with R/R neuroblastoma (either in the bone or bone marrow), in conjunction with GM-CSF, who have previously undergone prior treatments and have responded to it in the form of partial responses, stable diseases, or even minor responses (43-48). Naxitamab, under the tradename DanyelzaTM, was approved for medical use based on the findings of a phase 1/2 and a phase 2 clinical trial (clinical trial identifier: NCT01757626 and NCT03363373, respectively) in the both of which the safety and clinical efficacy of Naxitamab were investigated in R/R patients with high-risk neuroblastoma (43, 46). In reference to the NCT01757626 trial, in a group with 28 enrolled neuroblastoma participants unresponsive to prior induction therapy, ORR was measured as 78%, while the 24-month progression-free survival of the patients was found to be 50% (43, 46). Moreover, based on the findings from another patient group consisting of 35 individuals with salvage therapy-resistant neuroblastoma, ORR was measured as 37%, while the 24-month progression-free survival of the patients was found to be 36% in 30 subjects qualified for evaluation (43, 46). In reference to the other clinical trial (NCT03363373), a small portion of the findings have been presented at a congress in October 2020, according to which ORR was reported as 68% and complete response was achieved in 59% of the patients (22 subjects with high-risk neuroblastoma who had shown incomplete response to salvage therapy or those with refractory disease) (43, 46, 48). Of note, the tolerability and therapeutic efficacy of Naxitamab were investigated as a combination therapy with GM-CSF (43, 46, 48).
2.6. Dostarlimab
Dostarlimab (also known as TS-042, GSK4057190A, and Jemperli®) is a programmed death (PD)-1 receptor-targeting humanized IgG4k antibody (49-54). In April 2021, dostarlimab received accelerated FDA approval based on the data resultant from the GARNET trial (code: NCT02715284), a multicenter, multicohort, open-label trial involving patients with solid tumors in advanced stages (50). In 2020, Oaknin et al. reported the data from two independent expansion cohorts of patients with recurrent or advanced DNA mismatch repair deficient (dMMR) or proficient (MMRp) endometrial cancer (EC) (50). The dMMR or MMRp status of the enrolled patients was confirmed using immunohistochemistry (IHC) (50). Moreover, the disease of these patients had advanced after treatment with platinum-based antineoplastic agents (50). The dMMR ORR was reported to be 44.7% (46 out of 103 patients), and MMRp ORR was reported to be 13.4% (19 out of 142 patients) (50). Also, the complete response of dMMR and MMRp was reported to be 10.7% and 2.1%, respectively (50). Moreover, partial response of dMMR and MMRp was reported to be 34.0% and 11.3%, and dMMR and MMRp stable disease was reported to be 12.6% and 21.8%, respectively (50). Also, the 18-month duration of response (DOR) was also reported to be 79.2% and 61.3% for dMMR and MMRp, respectively (50). According to the mentioned data, Oaknin et al. (50) concluded that dMMR status is associated with a higher response rate.
3. Conclusion
Thirty-five years have passed since the first monoclonal antibody was granted FDA approval. As of May 2021, FDA has approved 100 monoclonal antibody products for medical use (55). As we discussed throughout this article, FDA-approved monoclonal antibodies for the discussed purposes have proven to be more efficient than most of the commonly available approaches. Monoclonal antibodies have also been a helping hand for other types of targeted cancer therapies, including CAR T cell therapy, immunotoxins, and bispecific T-cell engagers (BiTEs®), on their way to clinical success (2, 13, 31-33, 56-60). However, the road to success for monoclonal antibodies has always had its ups and downs. Many products face unpredicted adverse events or toxicities, which can vary from mild to severe. Such unfortunate happenings might halt a clinical investigation or may block the way of the associated product for upper-level investigations. However, the number of monoclonal antibodies receiving FDA approval is expected to increase in the upcoming decades.
