








1. Context
One of the most devastating diseases globally is cancer. It is a significant global health concern that affects people worldwide (1). Worldwide, cancer has consistently ranked as the leading cause of mortality (2). The ability of the body's immune cells to recognize and eliminate newly formed cancer cells when they are still few in number is likely more crucial to the development of cancer than the transformation of a healthy cell into a cancerous one (3). According to certain registries, the incidence of new cases of lung cancer in men is as high as 90 per 100 000, while in women, it is only 38 per 100 000. Women have the highest rates in North America and Northern Europe, while men have the highest rates in the USA and Eastern Europe. Apart from Turkey, no other nation has such high rates. Due to low survival rates, the mortality rate from lung cancer is typically equivalent to the morbidity rate, even in more industrialized countries (4). According to the Cancer Statistics Report 2020, the annual percentage increase in cancer diagnoses in India was 1. One to two percent between 2010 and 2019. Nationally, the number of deaths attributable to cancer increased by 0.11 percent over the same time frame. As a percentage of the population, women had a rate of 103.6 per 100 000, while men's rate was 94.1(5).
Lung cancer has claimed the lives of almost 2.21 million individuals globally, making it the world's second-biggest cause of cancer-related fatalities (6). Small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) are the two types of lung cancer, with NSCLC accounting for approximately 85 percent of cases. The two most prevalent histological subtypes of NSCLC are adenocarcinoma and squamous cell carcinoma. Alterations to the deoxyribonucleic acid (DNA) of the cancer cells allow for further molecular differentiation (7). Non-small cell lung cancer comprises a molecularly diverse population with a history of unsatisfactory responses to targeted therapy techniques, including downstream MEK inhibition, farnesyl transferase inhibition, and synthetic lethality screening (8). The details of the currently available therapy for NSCLC are provided in Table 1 below.
| Sl. No | Targets | Current Medication | Current Status of Approval |
|---|---|---|---|
| 1 | MET protein | Cristinib (9) | FDA approved (August 2011) |
| Cabozantinib (9) | FDA approved (April 2016) | ||
| MGDC265 (9) | Phase II study closed early | ||
| AMG208 (9) | Phase II study closed early | ||
| Altiratinib (9) | Pre-clinical study completed (2015) | ||
| Golvatinib (9) | Pre-clinical testing completed (2012) | ||
| 2 | EGFR mutations | Erlotinib (10) | FDA approved (November 2004) and modified as EGFR mutations specific (October 2016) |
| Gefitinib (10) | FDA approved (May 2003) | ||
| Icotinib (10) | SFDA, China approved (June 2011) | ||
| Afatinib (10) | FDA approved (July 2013) | ||
| Osimertinib (10) | FDA approved (April 2018) | ||
| W2015-S (11) | Pre-clinical testing completed (Jan 2021) | ||
| 3 | PD1/PDL1 expression | Nivolumab (12) | FDA approved (August 2021) |
| Pembrolizumab (12) | FDA approved (July 2021) | ||
| Cemiplimab (12) | EU approved (June 2019) | ||
| Atezolizumab (12) | FDA approved (March 2019) | ||
| Durvalumab (12) | FDA approved (February 2018) | ||
| Avelumab (12) | FDA approved (June 2020) | ||
| 4 | RET fusions | Pralestinib (13) | FDA approved (September 2020) |
| Selpercatinib (13) | FDA approved (May 2020) | ||
| HM06 (14) | Phase I/II completed (April 2022) | ||
| TPX-0046 (14) | Phase I/II undergoing (November 2022) | ||
| 5 | NTRK gene fusion | Larotrectinib (15) | FDA approved (November 2018) |
| Entrectinib (15) | FDA approved (August 2019) | ||
| 6 | BRAF mutations | Vemurafenib (16) | FDA approved (November 2017) |
| Dabrafenib + Trametinib Combination (17) | FDA approved (June 2017) | ||
| 7 | KRAS mutations | Sotorasib (AMG510) (18) | FDA approved (May 2021) |
| Adagrasib (MRTX849) (19) | FDA accelerated approval (December 2022) |
Currently Available Treatments for Various Targets of NSCLC
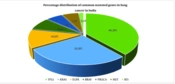
When attempting to distinguish between epidermoid carcinoma and adenocarcinoma, especially in tumors with weak differentiation, specific language and standards are used. This is due to the fact that lung cancer pathological classification is constantly changing. More recently, there has been a rise in the significance of correctly identifying the histological subtype of lung cancer due to the availability of an increasing number of therapeutic medicines designed to treat certain subtypes (20). According to Globocan 2020, out of a total of 1 324 413 cancer cases, India has reported 72 510 instances of lung cancer, which accounts for 5.5 percent of all cancer cases. Of these 72 510 cases, lung cancer in males accounts for 51 675 (71.26 percent) of the total lung cancer cases (21). A recent analysis of 230 lung adenocarcinoma cases offered a molecular profile. The most commonly mutated gene was TP53 (46%), followed by KRAS (33%), EGFR (14%), BRAF (10%), PIK3CA (7%), MET (7%), and RT1, a small GTPase (2%), and a set of tumor suppressor genes, comprising STK11 (17%), KEAP1 (17%), and NF1 (11%) as depicted in Figure 1 (22).

Graphical depiction of the percentage distribution of frequently altered genes in lung cancer patients in India
2. Targets for NSCLC
2.1. Mesenchymal-Epithelial Transition Protein Amplification
The mesenchymal-epithelial transition (MET) protein is the sole known receptor for the hepatocyte growth factor (HGF) and has recently been discovered as a potentially promising target in various types of human cancers, including NSCLC (23). The MET signaling system can be activated through various mechanisms depending on the context. Recent clinical data have shed light on the types of tumors and patient groups that could benefit from therapy using MET pathway inhibitors. Several drugs targeting MET have undergone clinical testing (24). In individuals with lung cancer, MET may be amplified, overexpressed, or activated (as evidenced by regulation of catalytic domain phosphorylation and juxtamembrane (JM) domain), or the gene may be mutated (in the JM domains or semaphorin) (25). Research on SCLC cell lines, for instance, has revealed that activation of the MET/HGF pathway during tumor colonization and growth serves multiple purposes (26). This occurs due to dysregulation of various biological processes, including cell growth and differentiation, transcriptional control, the G1/S checkpoint in the cell cycle, cytoskeletal functions, survival, and apoptosis (27). The tyrosine residues within the tyrosine kinase domain of MET regulate its kinase activity. Crucial for attracting downstream adapters such as the protein Growth Factor Receptor-Bound Protein 2 (GRB2) and the GRB2-associated binding protein 1 (GAB1) are the C-terminal regulatory tail tyrosine residues (28). The extracellular domains of MET interact with HGF to induce dimerization and transphosphorylation of tyrosine kinase residues in the cytosolic catalytic domain (29). Subsequently, autophosphorylation occurs, providing sites for the binding of adaptor proteins. This leads to the activation of several downstream pathways crucial in cancer mechanisms, including PI3K, MAPK, and STAT3, among others (30). The adaptor protein GAB1 increases the number of sites available for signaling molecules to bind (31).
Pennacchietti et al. (as cited by Sadiq and Salgia) discovered that MET is overexpressed in hypoxic tumor areas. Hypoxic activation leads to transcriptional regulation of the MET proto-oncogene, increased MET levels, and HGF signaling. Blocking the expression of the MET gene halts the proliferation of invasive cells induced by hypoxia. Evidence suggests that the Wnt pathway regulates MET expression in colon cancer. B-cell expansion relies on the nuclear transcription factor PAX5, which is absent (32).
Altiratinib, cristinib, MGCD265, cabozantinib, AMG208, altiratinib, and golvatinib are all multikinase MET protein inhibitors. Tepotinib (MSC2156119J), capmatinib, and tivantinib are examples of adenosine triphosphate (ATP)-competitive and ATP-noncompetitive selective MET inhibitors, respectively (9).
2.2. EGFR Mutations
EGFR was the first member of the RTK family to be discovered. The EGFR family of RTKs comprises four distinct receptors: EGFR (ErbB-1/HER1), HER2 (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4) (33). These RTKs feature a single hydrophobic transmembrane domain (TM), an extracellular ligand-binding domain, and a cytoplasmic tyrosine kinase domain (34). EGFR was the first member of the RTK family to be identified as a prototypical member (35). It becomes active upon forming homodimers and heterodimers with various external growth factor ligands, such as transforming growth factors (TGF) and EGF (36).
Targeted treatment is ineffective for patients with EGFR-TKI-resistant lung cancer. Activation of STAT3, a major modulator of various cellular activities including cell multiplication, differentiation, immunological function, and angiogenesis, is a significant factor in drug resistance for oncogene-driven malignancies in targeted therapies (37). Stimulated STAT3 regulates immune checkpoint proteins and tumor environment cytokines (38). Targeting STAT3 holds therapeutic potential and could assist TKI-resistant lung cancer patients in various ways (11).
Because of the development of resistance systems, upcoming treatment regimens and the next generation of EGFR inhibitors may probably broaden to more specifically target the specific co-mutations (39). It is interesting that several mutations that aid in resistance to Osimertinib are still somewhat responsive to 2nd generation EGFR inhibitors (40). These medications may provide an instant answer for a subset of patients, even though their action against native EGFR and the resultant side effects restrict their widespread human usage (41).
Erb-B2, a tyrosine kinase receptor, is expressed by the HER2 gene. It activates the MAPK and PI3K pathways in various ways to overcome EGFR TKIs like lapatinib, hence causing resistance (42). Patients with no T790M mutations who progressed on first-generation EGFR TKIs had an increased frequency of HER2 amplification in their tumor tissues (43). In the same way that different platforms report varied criteria for defining MET amplification, NGS techniques have been established with varying criteria for defining HER2 amplification based on reports from various platforms and validation cohorts, which may include tissue or plasma samples (44). Multiple mutations that are active take place inside the exons numbered 18 to 21 have the potential to cause abnormal regulation of EGFR activity (expressing the kinase domain) (45). Exon 19 mutations, which are characterized by the replacement of leucine for arginine, account for 40 - 45 percent of majority of EGFR mutations even though exon 21 mutations account for 45 percent of all EGFR mutations and include the deletion of amino acids 747 - 750 (46).
The EGFR mutation test is performed on DNA isolated from a tissue sample taken via a biopsy of the tumor (47). It is generally accepted that tissue biopsy serves as an inadequate gold standard or reference standard (48). The EGFR T790M mutation may be absent in certain cancer areas due to the heterogeneity of the cancer (49). Patients may have an advanced stage of NSCLC if their condition progresses throughout the course of the disease (50). Patients who undergo tissue biopsies thus have a higher chance of unfavorable results than those who receive cell-free circulating tumor DNA (ctDNA) blood testing (also called liquid biopsy) (51). The median progression-free survival (mPFS) for first-generation TKIs (icotinib, gefitinib, and erlotinib) or second-generation TKIs (afatinib) was reported to be between 9 and 13 months, whereas the mPFS for third-generation EGFR TKIs (osimertinib) was 18.9 months. Therefore, EGFR TKI is the current standard of treatment used as the first line of defense for these patients (52). Nevertheless, EGFR TKI resistance is inevitable, and the development of novel treatment techniques to use when it occurs is still a problem that has not been overcome (10).
Recently, Zheng Q et al., who studied the sensitization of resistant NSCLC, concluded that W2014-S, a molecule isolated from their own in-house library, successfully sensitized the resistant NSCLC cell lines and prevented the development of PDX tumor xenografts and human NSCLC cell xenografts in mice (11).
2.3. Programmed Death-1/ Programmed cell Death 1 Ligand 1 Expression
Programmed death-1, also known as PD-1, is an immunological checkpoint that prevents autoimmunity and controls immune responses that are excessive in response to antigens (53). Natural killer T (NKT) cells, B lymphocytes, T lymphocytes, activated monocytes, and dendritic cells are some of the immune cells that possess the capacity to express PD-1. Dendritic cells also have the potential to express PD-1 (54). The manifestation of programmed cell death 1 ligand (1PD-L1) functions as the main immune suppressive mechanism in NSCLC via the interaction of the PD-1 and PD-L1 axis (55). PD-L1 can behave as a coregulatory wave by attaching to the repressive PD-1 receptor after T cells have been activated by major histocompatibility complex (MHC) peptide-based antigen recognition and T cells have been activated. This eventually results in the downregulation of lymphocytes and other immune cells (56). This occurs when T cells are activated in response to an antigen through a contact between their T-cell receptor and a peptide from the MHC as shown in Figure 2 (57). The inhibition of T cell proliferation and effector activities may be achieved by the attachment of PDL1 or PDL2 to the PD1 protein found on the cell's surface (58). PDL1 is found in many immune cells as well as non-immune cells and is elevated by pro-inflammatory mediators like interferon (IFN) and Interleukin 4 (IL4) via signal transducer and activator of transcription 1 (STAT1) and IFN regulatory factor 1 (59). A recent randomized control trial study in volunteers with PDL1-positive NSCLC discovered that PD1 inhibitor + pembrolizumab was well-endured and enhanced overall and PFS in volunteers when compared to pembrolizumab treatment alone (60). Notably, the results of the experiment showed that the inclusion of NK cell treatment was not linked with any negative side effects (61).

Expression of PD-1/PD-L1 and its role in the development of tumors
MicroRNAs are small nucleotide molecules that regulate cancer cell proliferation, migration, invasion, and apoptosis during tumor formation. miR-155, miR-34, miR-33a, miR-21, miR-873, and miR-146a affect PD-1/PD-L1 expression in human carcinoma (62). They alter human immunology. miR-34 inhibits PD-L1 in NSCLC (63). Sulfamethoxine and sulfamethimazole antibiotics, aptamer-drug conjugates like pegaptanib, monoclonal antibody treatments like nivolumab, pembrolizumab, and cemiplimab, and atezolizumab, Durvalumab, and avelumab targeting PD-1 and PD-L1 are examples of PD-1/PD-L1 checkpoint inhibitors (12).
2.4. RET Fusions
Four distinct research teams from Korea, Japan, and the US separately reported the recognition of the first RET fusion in lung tumor in 2012, which was known as KIF5B-RET (64). One of the groups looked at tissue and body fluid samples from a lung cancer patient who had never smoked and was 33 years old (65). The patient had adenocarcinoma in both lungs. The EGFR mutation, the KRAS mutation, and the EML4-ALK fusion gene were the three renowned driver alterations in lung cancer at the time (66). The patient tested negative for all three of these mutations in their cancer (67). It is hypothesized that an additional therapeutic target for treatment of LADC might be found in the RET gene (68). Only one to two percent of LADCs will have the RET fusion (69). Inhibitors of RET tyrosine kinase that are already on the market and have been licensed by the FDA in the United States have shown positive therapeutic benefits in vitro, in vivo, and in a small number of patients (70).
Mesenchymal-Epithelial Transition amplifications and solvent front mutations impede RET fusion (71). RET resistance mutations were rare in this population, and gatekeeper variants targeting the V804 residue did not arise (72). Despite pralsetinib and selpercatinib's preclinical success against RET V804 mutations, larger cohorts are required for validation of the RET mutations' low incidence and narrow scope (13). In two selpercatinib-resistant patients and one patient with a cis mutation, Tan et al., discovered RET V804 and G810 mutations 71. Despite preclinical findings, further study is required to determine if gatekeeper mutations may impart resistance to selpercatinib and/or pralsetinib and if the RET resistance mutation spectra (and non-RET resistance variations) vary amongst the two drugs (73). The RET inhibitors of the next generation, HM06 and TPX-0046, are now participating in Phase I/II clinical trials, which are being suggested for the therapy of individuals who have RET-altered advanced solid tumors (14).
2.5. NTRK Gene Fusion
Tropomyosin receptor kinases (TRKA, TRKB, and TRKC) are the proteins encoded by the NTRK alleles (NTRK1, NTRK2, and NTRK3) respectively. These NTRK genes bind to ligand neurotrophic factors such as nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophin factor to convey signals into the cell (74). This aids nerve cells in both the central and peripheral regions to develop and persist (75). Somatic mutations in NTRK have been identified in various tumor types, including acute myeloid leukemia, large-cell neuroendocrine carcinoma, NSCLC, and colorectal cancer (76). For instance, mutations in NTRK2 affecting TRKB have been discovered in the NCI-H2009 lung cancer cell line at two distinct kinase domain regions (T695I and D751N), within the extracellular region (L138F), and near the kinase domain (P507L) in colorectal cancer patients (77). TRKA and TRKB homologs lacking this insertion can only be significantly activated by NGF and BDNF, respectively (78). In contrast, due to this insertion, the TRKB splice variant is readily activated by NT-3 and NT-4 in addition to BDNF, while the TRKA splice variant is activated by NT-3 in addition to NGF, both effects dependent on the presence of the insertion (79).
Raising awareness about this biomarker is crucial to employing a therapeutic approach that is highly effective and well-tolerated in treating both common cancers like NSCLC, where extensive tumor genotyping has become clinical practice, and rare tumors, where NTRK rearrangements are pathognomonic (80). Compared to other histological types, especially adenocarcinoma, NTRK1 and NTRK2 are more frequently expressed in epidermoid carcinoma (81). In a cohort of 686 individuals with epidermoid carcinoma, NTRK1 showed high specificity (92.8%) but mediocre sensitivity (71.6%). NTRK2 exhibited a specificity of 96.4% in the same cohort, with sensitivity at just 51.3%. NTRK2 is a potential biomarker for squamous cell proliferation based on several studies (82). Larotrectinib and entrectinib, two novel cancer treatments developed to target NTRK-associated tumors and approved by the FDA in 2018 and 2019 respectively, exemplify the advancements enabled by this research (15). Larotrectinib, a selective pan-Trk inhibitor, was the first oral "tumor-agnostic" medication and the first drug developed for both adults and children, also being the first treatment administered orally (83).
2.6. BRAF Mutations
The RAS-RAF-MEK-ERK axis regulates cellular growth, with the BRAF gene playing a role in this axis (84). The BRAF gene codes for a serine/threonine kinase (85). There is no correlation between ethnicity and the incidence of lung cancer caused by BRAF mutations, which range between 1.5 and 3.5 percent (86). According to various investigations, BRAF mutations may be found in between three and five percent of patients with non-squamous NSCLC (87). BRAF encodes a serine/threonine-protein kinase that affects cell development. BRAF's kinase domain is encoded by amino acids 457 - 717 (88). The activation loop of the kinase interacts with the phosphate-binding loop, locking it (89). If the activation loop is phosphorylated, BRAF can phosphorylate and activate the MAPK ERK1/2 signaling route (also known as MEK1/2). ERK1/2 phosphorylates MAPKAPKK and cytoskeletal proteins including vimentin and keratin-8. ERK 1 and 2 activate transcription factors such as FOS, TP53, and ELK1(90, 91). Human cancers have around 200 BRAF mutations categorized based on kinase activity, RAS-dependence, and dimerization status (92). Class 1 BRAF mutations (V600E/D/K/R) are more common in solid tumors; they activate BRAF kinase and the MAPK pathway. Class 2 BRAF mutations signal as constitutively active dimers, depending on MAPK pathway activation (93). RAF inhibitor resistance exists for class 1 and class 2 BRAF mutations that are RAS-independent (94). Class 3 BRAF mutations are RAS-dependent and have little or no kinase activity (95). Since these mutations require upstream MAPK activation and are not independent drivers, they often occur with RAS mutations or NF1 deletion. Blocking upstream RAS signaling might be therapeutic (96).
One-arm cohort research by Subbiah et al. In total, 62 BRAF V600-mutated NSCLC patients (61 with the V600E mutation and 1 with an unexplained V600 mutation) were included, and eight (13%) of them had never received any therapy before. Three patients (4.8%) had CNS metastases, and the majority of patients (n = 36, or 58 percent) had quit smoking. Most patients (n = 58, or 94 percent) had been diagnosed with adenocarcinoma. Platinum agents, pemetrexed, and taxanes were the most frequently used previous chemotherapies (39 of 54 patients; 72% of total), with two prior systemic regimens being the median number. The median follow-up period for this study was 10.7 months (IQR, 4.3 to 17.1 months). Seven patients (11%) discontinued vemurafenib for a variety of reasons, including progressive disease (in 41 of 62 patients, or 66 percent), adverse events (in 6 of 62 patients, or 10 percent), death (in 4 of 62 patients, or 6 percent), patient withdrawal (in 2 of 62 patients, or 3 percent), or physician decision. Six patients transitioned into an expansion study, and one patient withdrew from the trial (in 2 of 62 patients, or 3 percent) (16).
Clinical studies on the first BRAF V600E-mutated NSCLC patients demonstrated potent anticancer benefits of the BRAF inhibitor dabrafenib combined with the MEK inhibitor trametinib (17). Non-small cell lung cancer patients with BRAF V600E, a potential resistance mechanism to EGFR TKI therapy, have few therapeutic options, and it was unclear how well dabrafenib and trametinib + osimertinib work as a combination (97). In two instances where the BRAF V600E mutant was most probably an acquired osimertinib-resistance mutation, the primary clinical decline during therapy occurred in one patient who already had brain metastases at the time of enrollment. There must have been a unique resistance mechanism in the brain metastases, or the medicine must have had trouble penetrating the blood-brain barrier (98).
2.7. KRAS Mutations
KRAS is the most widespread oncogene in NSCLC (99). In contrast to other significant mutational subgroups of NSCLC, research conducted in preclinical settings lends support to the concept that KRAS mutations may be susceptible to immunotherapy techniques (100). This effectiveness is particularly connected with the presence of TP53 co-mutation (8). The synergistic effect of KRAS and TP53 co-mutation boosts the synthesis of neoantigens produced by lung cancer cells, which should, in theory, provoke an inflammatory response within the immunological setting of the tumors (101). Whenever there is a KRAS mutation, modifications to LKB1 induce the first line of defense against programmed cell death protein 1 blocking, which makes these tumors resistant to immunotherapy (102). GFPT1 suppression does not alter NSCLC cell viability, even though both catalyze HBP entrance (103). KRAS/LKB1 mutant NSCLC depends on GFPT2 and associated pathway components. Azaserine reduces KRAS/LKB1 co-mutant cell viability (104). Oncogenic KRAS and LKB1 deletion predict aggressive cancer in mice and humans. Oncogenic KRAS's loss of LKB1 impacts glutamine flow via GFPT2, HBP's rate-limiting phase. GFPT may boost glutaminase's function in anaplerosis by replenishing glutamate (105). The detailed mechanism of KRAS mutation and formation of tumors has been shown in Figure 3. According to Judd et al., 4,706 (17,095) NSCLC samples had a KRAS mutation. The most prevalent were G12C (40%), G12V (19%), and G12D (15 percent). There was no age difference, although female patients had higher KRAS mutations (31.35 vs. 23.7%). KRAS mutations were seen in samples of squamous cell carcinoma (4.4%) and adenocarcinoma (37.2%). 1,841 patients smoked never, sometimes, or now, which is incompatible with the biology of KRAS. 43% of G12C, 7% of G12A, and 8% of G12D patients smoked. Compared to G12A patients, 16% of G12D patients never or lightly smoked. G12C mutation smokers were 43% (106). Clinical studies for sotorasib (AMG510), the first small molecule inhibitor of KRAS (G12C), have begun (NCT03600883). KRAS (G12C) protein is rendered inactive by binding to Cys12 of the inducible S-IIP (18). Compared to ARS-1620, performance is increased by a factor of ten when AMG510 is used in conjunction with the distinctive surface groove generated by an alternative orientation of His95 on KRAS (107). The FDA authorized AMG510 in May 2021 as the first therapy for patients with KRAS (G12C)-mutant NSCLC who had previously received at least one prior systemic drug (108). The United States Food and Drug Administration has designated adagrasib as a breakthrough medicine for the treatment of non-small cell lung cancer in the KRASG12C subtype (19).

Mechanism of KRAS mutation in Lung cancer formation
3. Discussion and Conclusions
Non-small cell lung cancer is one of the two lung cancer subtypes that has been extensively studied. However, due to challenges such as changes in protein amplifications, mutations, and co-mutations, the mechanism of tumour development is constantly evolving, highlighting the importance of staying updated on lung cancer therapy. Conversely, due to its poor prognosis, SCLC is not studied as extensively.
According to the findings of this study, certain co-mutations, such as EGFR-MAPK co-mutations and KRAS-TP53 co-mutations, render the target unresponsive to currently available treatments. Additionally, other challenges, such as specific protein amplifications, may be identified during the prognosis of lung cancer. These protein amplifications have the potential to serve as targets for lung cancer medication. Figure 4 summarizes the various drugs targeting specific sites to control tumor growth.

Schematic Diagram of the Mechanism of Lung Cancer and Its Target-Specific Inhibitors
