

1. Introduction
In the 21st century, the medical facilities and avenues have advanced to extents where researchers now speak of DNA and RNA vaccines. Such advancements were not available in previous centuries. Although war still exists in the 21st century, with advanced medical treatment, the casualties are lower in military forces. Every century has witnessed its own great war. The 20th century witnessed two world wars. In both World War I (WWI) and World War II (WWII), thousands of soldiers died of cholera, pneumonia, influenza, typhoid, mumps, tetanus, and rabies, besides other fatal infections. Wars dated prior to the WWI and WWII witnessed millions of soldiers’ deaths due to infections from battle wounds and unhygienic conditions in trenches.
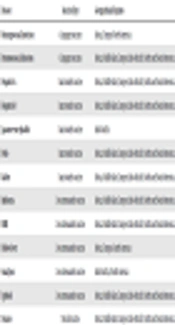
According to statistics, the majority of the fatalities did not occur due to the combat but due to the disease (1). The infections are transmitted through mosquitoes, ticks, and urine of rodents or are blood-borne, water-borne, or soil-borne. Even the consumption of unpasteurized dairy products, undercooked meat, or the use of animal feces are the factors that may lead to infection. Additionally, there are other routes such as exposure to animal-derived products and hides, as well as sexual contact. In modern warfare, the soldiers are trained in a six-component approach comprising preparation, education, personal protection measures, vaccination, chemoprophylaxis, and surveillance (1). Military forces are vaccinated prior to service. They are recommended taking vaccines for diseases mentioned in Table 1. A few vaccines can be omitted from the inventory based on the region (2). Vaccines for anthrax, smallpox, and yellow fever are usually region-specific in nature. Further, vaccines for diseases such as pneumococcal and meningococcal infection, tetanus, rabies, and influenza require booster doses every five years (3). Hence, an emphasis has been laid on increasing the longevity of immunization, as well as the reduction of side effects such as fever and body ache after dose inoculation. While DNA vaccines and recombinant vaccines for most diseases are in the clinical trial phases, researchers have sought to improve the well-established traditional vaccines. This article aims to highlight the techniques used for producing particular types of vaccines.
Table 1.Vaccines Given to Military Forces Based on the Geographical Location of Their Posting
| Disease | Vaccine Type | Geographical Regions |
|---|---|---|
| Meningococcal infection | Conjugate vaccine | Africa, Europe, North America |
| Pneumococcal infection | Conjugate vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
| Hepatitis A | Inactivated vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
| Hepatitis B | Inactivated vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
| Japanese encephalitis | Inactivated vaccine | Indo-Pacific |
| Polio | Inactivated vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
| Rabies | Inactivated vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
| Influenza | Live attenuated vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
| MMR | Live attenuated vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
| Yellow fever | Live attenuated vaccine | Africa, Europe, South America |
| Small pox | Live attenuated vaccine | Indo-Pacific, North America |
| Typhoid | Live attenuated vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
| Tetanus | Toxoid vaccine | Africa, Middle East, Europe, Indo-Pacific, North and South America |
2. Live Attenuated Vaccine
Live attenuated vaccines (LAVs) provide immunity to individuals against a specific disease as they provide antigenic memory in the form of attenuated pathogens to the host’s immune system. LAVs allow the host immune system to produce primary lymphocytes for the antigens displayed by these attenuated pathogens. The production of primary lymphocytes is time-consuming as the specificity for a particular disease is initially absent in the immune system. Thus, LAVs provide the immune system with an opportunity to recognize and retain the memory of the antigenic specificity for the inoculated virus (4). The mode of action of the vaccine is slow approximately; hence, adjuvants are not required to further increase the duration of immunity.
In the past, LAVs were producing by continuous passages and selections. However, the empiric methods of production would sometimes lead to the reversion of the attenuated virus into a wild lethal strain (5). Rational methods have been developed over the decades for the production of attenuated pathogens as they reduce the probability of reversion of the virus into a wild strain (6). The strategies for development are listed in the following.
2.1. Attenuation by Loss of Genetic Pool
In the case of viral pathogens, the population is not a single genotype but a mixed genotype due to rapid mutations in the viral RNA; hence, it is essentially a “quasispecies”. This quasispecies is necessary for the survival and virulence of the pathogen as it provides the pathogen with a better chance of survival when infecting an individual by granting the virus to have the ability to adapt to a new environment when infecting a host (7). Degradation in this quasispecies can be brought about by propagating the virus in an atrophic host, resulting in the loss of genetic diversity for successful infection of the trophic host (8). The oral poliovirus vaccine was produced by limiting the quasispecies of poliovirus by conducting several passages of Mahoney type-1 strain and Saukett type-3 (9) strain in rats and mice with subsequent passages in cell cultures.
The vaccine for smallpox, which was obtained from milkmaids suffering from cowpox, can be considered the first-ever vaccine made using this concept (10-12). The variola virus had not been inoculated in another host; however, it can be deemed that cowpox was an already existing attenuated model of smallpox present in cows that would mildly infect humans. Thus, when Edward Jenner inoculated James Phipps with cowpox pathogen, followed by variola virus, James survived without developing smallpox (10, 11, 13).
The virulence of a pathogen would decrease if the overall genotypic diversity of the population was restricted by subjecting the viral population to continuous genetic variation and competition (14). For example, the measles vaccine is produced by inoculating and passaging the pathogen in chicken embryonic fibroblast, resulting in the attenuation of the virus (7). The vaccine for yellow fever was produced in a similar manner. Two strains of yellow fever were independently produced by different groups. The yellow fever-French neurotropic vaccine (YF-FNV) was produced by taking a wild strain and passaging the strain 128 times in the intracerebral mouse brain (15) while the Asibi strain for yellow fever was produced by another wild strain passaged in the mouse culture, followed by chick embryo tissue (16, 17). The Asibi strain was attenuated by passaging in HeLa cells in the previous decade (18). An experiment on the prolonged cultivation of yellow fever virus in vitro demonstrated that the virulence decreased for the cultivation in chick embryo tissue while the virulence was retained in the prolonged cultivation in mouse tissue (17).
The controversial Urabe strain for mumps, whose use stopped as a vaccinating strain because of developing meningitis and encephalitis (19), was produced by attenuating the pathogen in chick fibroblast (5, 20). This is while the Jeryl Lynn strain of mumps is produced owing to this concept by inoculating it into specific pathogen free (SPF) chicken embryonic fibroblasts (21) and cell cultures of chick embryo (20, 22). Strains such as rubini strain (23) are obtained by isolating the pathogens from the patient, followed by SPF chicken fibroblasts and MRC-5 cells passages (16, 24). The Leningrad-3 strain is a combination of five strains of mumps while L-Zagreb strain is a further sub-cultivation of the Leningrad-3 strain in chicken fibroblasts (25, 26).
The influenza vaccine is produced using a similar strategy wherein the dominant strains of influenza A (H1N1 and H3N2) and influenza B are collected for the next infective season (27) and propagated in embryonated chicken eggs of typically 2-weeks-old (28-30). Since such large-scale production of single-use bioreactors is not possible to meet the demands for bulk production of the vaccine, the host is changed to a mammalian cell line, which is typically the Vero cell line or the MCDK cell line in the case of Flucelvax that is produced by Novartis (31).
In the case of bacterial pathogens, a synthetic quorum sensing environment that induces the expression of attenuated factors can be constructed and induced on the population of the pathogen (32). Thus, the virulence of the pathogens decreases by inducing a bottleneck to prevent the expansion of genetic diversity of the population. The attenuation of a pathogen can be confirmed by checking the markers for virulence in the pathogen’s genome sequence. This indicates that there are certain genes responsible for the reduced virulence of a pathogen. Hence, by targeting genes and “switching them on or off”, a virulent pathogen can be engineered to be non-virulent and in the case of vaccines, it can be engineered to be attenuated. These engineered pathogens are attenuated in nature either due to a low replication rate or due to a decrease in the expression of toxins or both (33). Polymerase-based attenuation relies on the mutation or replacement of a specific residue on the pathogenic RNA-dependent RNA-polymerase (RdRp) such that the genetic variation of the quasispecies is lowered. Site-directed mutation mediated through PCR overlapping is used to mutate a specific residue (34). The mutation aims at increasing the fidelity of RdRp. By increasing the fidelity, the number of mutations occurring reduces; hence, a loss in genetic variation is evident (35). This method reduces the probability of the attenuated virus from becoming a wild type even after several passages, thus effectively reducing the chance of reversion.
2.2. Attenuation by Codon Targeting
A codon is degenerate in nature, that is, there are multiple codons coding for the same amino acid. In an organism, the genome has degenerate codons; however, it has been observed that among synonymous codons, there are certain codons that are expressed more; thus, a codon bias may exist in certain species (36, 37). For example, in the case of poliovirus, a change in the genome sequence of type-2 strain by increasing the frequency of CpG and UpA dinucleotides results in the decrease of codon bias, followed by the attenuation of the virus due to the lowered expression of toxins and lowered replication rate, which were found to be conserved after several passages in HeLa cells (36, 38, 39).
Deoptimized codons have several advantages complying with the strategy of vaccine development. First, the deoptimized codons express proteins that are identical to the wild type and the attenuated type but the translational efficacy is greatly reduced. Hence, the antigenicity is not affected and the immune response elicited is similar to the response toward a natural infection. Second, this technique can be applied to a large number of viruses as it is a systemic approach rather than an empiric one. Finally, the point mutations are brought about in thousands of synonymous codons, thus minimizing the possibility of reversion to a wild lethal strain.
2.3. Attenuation by Auxotroph
Auxotrophic mutants of a strain are incapable of producing a naturally synthesizable compound by the normal strain, thus requiring the naturally synthesizable compound to be additionally present in the media for normal functioning of the auxotrophic strain. Auxotrophic strains are usually mutated by deleting or silencing a gene that is responsible for the production of a growth-linked product. As a direct result of this deletion, the mutant strain is unable to proliferate at a rate similar to that of the normal strain.
In LAVs, the use of auxotrophic mutants is possible due to the fact that the mutant is unable to receive sufficient quantity of metabolites required for growth inside the host system. To make the vaccine more effective and less revulsive, the strain is further manipulated to decrease the invasive and toxin production capability. A successful model of such a vaccine can be observed for S. typhi. The hypoxanthine, thiamine, and adenine auxotroph of S. typhi (40) constructed through two deletion mutations from the Ty2 and CDC 10 - 80 strain has been reported as a possible candidate for the live-oral vaccine for typhoid (41).
3. Inactivated Vaccine
Unlike LAVs, inactivated vaccines provide immunity to the host by inducing an immune response in the host toward the injected virus that has been killed or inactivated via chemical or thermal means such that it cannot further replicate or survive in the host organism. Several novel agents and methods of inactivation have been described (42) such as ascorbic acid used for preparation of the rabies vaccine (43), psoralen-induced inactivation for the dengue vaccine (44), UV treatment and Gamma irradiation for the inactivated influenza A vaccine (45, 46), the use of ethylenimine derivates, heat inactivation for poliovirus (47), formaldehyde, β-propiolactone, and so on (48, 49). The rabies vaccine is produced by binary ethylenimine after propagating the virus in BHK cells; however, vaccine inactivation by this method is less stable (50). Nonetheless, formaldehyde and β-propiolactone (BPL) are widely used for the production of inactivated vaccines because of higher efficiency and stability retention. Inactivated vaccines are second to LAVs in stimulating and mimicking the immune response to a natural infection; however, the immune response generated by inactivated vaccines is weaker than the immune response generated by LAVs, thus requiring “booster” injections and immunological adjuvants to provide a stronger immune response against the pathogen.
Inactivated vaccines are stable and can be easily maintained when compared to LAVs. Moreover, inactivated vaccines can be used in multivalent combinations to provide immunity against different strains and viruses in a single dose. Inactivated vaccines can be further classified as whole virus vaccines that contain completely killed/inactivated virus, split virus vaccines that contain disrupted viruses using a detergent to make the split virus vaccine, and subunit virus vaccines produced by only purifying out the antigen from the virus such that the purified antigen can mimic the stimulation of a natural immune response.
3.1. Inactivation by Thermal Treatment
Inactivation by heat treatment is the simplest technique that can be used for inactivating viruses. Thermal inactivation is followed by chemical inactivation that may result in increased vaccine toxicity (51). For unknown viruses, the virus sample is usually heated below 100°C for a long duration or over 100°C for a short duration. The thermal inactivation point determines the lowest temperature that would suffice to inactivate an unknown virus when treated for 10 minutes. The temperature is increased at intervals of 10°C from the first exposed temperature and the interval is reduced to 5°C when inactivation first occurs (52). A common drawback of thermal inactivation is the denaturing of RNA and DNA strands, as well as proteins, resulting in ineffective vaccines. Despite this drawback, both hepatitis A and hepatitis B vaccines are produced by heating the virus to 56°C for 30 minutes (53). A study demonstrated that heat inactivation at a temperature of 65°C for a period of 15 minutes (54) was sufficient to completely inactivate poxvirus, picornavirus, toga virus, coronavirus, orthomyxovirus, rhabdovirus, herpes virus, lentivirus, and retrovirus while parvovirus and Papovaviruswere inactivated with heat treatment for 90 seconds at 103°C (55).
3.2. Inactivation by Formaldehyde
Formaldehyde is the simplest aldehyde that usually acts as a reducing agent unless a stronger reducing agent is added to the reaction mixture. It brings about various modifications such as methyl groups, methylene bridges, and Schiff bases in proteins. This results in the inactivation of proteins.
Generally, formaldehyde stock solutions are diluted to a final concentration of 0.4% formalin. This concentration requires an inactivation period of up to three weeks for higher titers of the virus at an incubation temperature of 2°C to 7°C. However, to inactivate more potent batches of the virus, the concentration of formalin is lowered to approximately 0.025% - 0.012% and the temperature is increased to 37°C - 40°C as used in the preparation of Salk’s vaccine (56). Therefore, the higher the concentration of formalin and the higher the temperature, the faster the rate of inactivation; however, the loss of immunogenicity might be observed due to the degradation and destruction of the toxin. Thus, the inactivation time should be optimized such that the immunogenicity is not lost while assuring the complete inactivation of the virus. Once inactivation of the virus is done, residual formalin is removed using sodium bisulfite (48).
Japanese encephalitis virus (JEV) is cultured in Vero cells and inactivated in formalin, followed by purification to obtain the antigens. The genetic analysis of all JEV isolates confirmed that they comprised a single serotype (57). This information is valuable for vaccine design (58). A study of a strain obtained from Vellore, India, demonstrated that inactivation by formalin at 22°C was faster and cheaper than inactivation by formalin at 4°C (59).
Formalin-inactivated hepatitis A virus showed an appropriate immune response in a study (51), thus, suggesting vaccine development by formalin inactivation (60). The inactivated poliovirus vaccine is produced by inactivation of three strains of poliovirus, Mahoney type-1, MEF-1, and Saukett type-3, using formaldehyde; however, a failure in the inactivation of the Mahoney strain due to resistance to formaldehyde resulted in the replacement of Mahoney strain with Brunenders strain type-1 (9).
In the case of live attenuated strains of influenza, including two subtypes of influenza A (H1N1 and H3N2) and two antigenically distinct lineages of influenza B, obtained from serial passages in embryonic chicken eggs or Vero cells depending on the host used, when further treated with formalin (61), it was observed that the surface glycoproteins hemagglutinin (HA) and neuraminidase (NA) characteristics were retained while effectively killing the cells, resulting in the formation of an inactivated vaccine (29, 62). Hence, this multivalent vaccine is a whole virus vaccine in nature that contains toxins from more than one strain (28). This whole virus vaccine when treated with Triton-X100 results in the production of the split virus vaccine (63).
3.3. Inactivation by β-Propiolactone
β-propiolactone (BPL) is a four-ringed lactone and is highly reactive toward nucleophiles. BPL is stable in concentrated liquid forms but readily degrades in aqueous solutions due to hydrolysis into non-toxic and non-carcinogenic products. This results in the complete elimination of BPL from the reaction mixture, thus eliminating the requirements of techniques for further removal of BPL from the product. Thus, BPL poses an advantage over formaldehyde inactivation methods as residual formalin has to be removed from the product. However, if excess BPL is present, it has to be neutralized using thiosulphate. The inactivation time of viruses is shorter when BPL is used than when formaldehyde inactivation methods are employed. Moreover, the temperature used during inactivation is low, thus reducing the risk of denaturation of epitopes due to thermal degradation.
BPL is used for the inactivation of the rabies virus. The PM strain and Flury HEP strain were adapted in WI-38 for several passages and a viral pool was prepared in the BHK cell line. These strains were then chemically inactivated by BPL at the 0.025% concentration at 4°C for various lengths of time (64). The PV/VERO-Paris strain of rabies was inactivated by BPL after culturing in Vero cells (65).
However, BPL directly interacts with nucleic acids to inactivate the virus. DNA and RNA are irreversibly alkylated and acylated by BPL, specifically reacting with N-7 of guanosine and N-1 of adenosine to some extent. Due to this modification, Gp is misread as Ap by the polymerase, resulting in numerous irreversible point mutations in the genome of the virus, hence rendering it inactive. Therefore, the proteins expressed are altered due to this nucleic mutation. Moreover, it has been observed in a study that BPL interacts with 9 amino acids, hence directly altering proteins. A loss in immunogenicity occurs due to these alterations. Unlike formaldehyde inactivation, the concentration and the temperature of BPL vary based on the virus being inactivated.
3.4. Inactivation by Psoralen
Treatment of viruses by psoralen is a relatively mild method to inactivate the virus. 4’-aminomethyl 4,5’-8-trimethylpsoralen in an inert environment along with UV radiation has shown to be an effective inactivator for the bluetongue virus (66) and immunodeficiency viruses (67). Psoralen inactivates viruses by intercalating between the base pairs of double-stranded nucleic acids, hence inhibiting the replication of DNA. The viral protein structure is preserved after inactivation and no residual toxicity is observed; however, inactivation by psoralen is not cost-effective (68).
4. Toxoid Vaccine
Toxoid is a bacterial endotoxin with suppressed properties by using chemicals such as formalin but maintaining the immunogenicity. On vaccination, the immune response is generated by the host body and the immunological memory is built against the molecular markers of toxoid but there is no manifestation of the disease in the host. However, no strong immune response is elucidated, necessitating booster doses.
The preparation of toxoid is done by the inactivation of endotoxins by chemical methods including oxidation such that the process is irreversible. The oxidizing agents could be either organic or metallic in nature. Organic oxidizing agents including hydrogen peroxide, aldehydes, sodium peroxides, N-chloro-4-methyl-benzene-sulfonamide sodium salt (chloramine-T), performic acid, dioxane peroxide, periodic acid, sodium permanganate, and sodium hypochlorite (69) are used primarily. The most preferred oxidizing agent is hydrogen peroxide as it is easily handled, easily available, and cost-effective (69). Aldehydes such as glutaraldehyde or formaldehyde form Schiff bases. Schiff bases are chemically unstable and may lead to reversible reactions leading to toxoid conversion into an active toxin (70).
A recent method involved the addition of small amounts of metal ions to oxidizing agents for the treatment of the toxin. Metal ions will chemically inactivate the toxin while maintaining immunogenicity. Oxidizing agents oxidize amino acids such as cysteine, cystine, methionine, tryptophan, or tyrosine at specific positions in the peptide (69). For example, tetanus disease is caused by endotoxin secreted by Clostridium tetani bacterium. The endotoxin from Clostridium tetani is isolated using a purifier (71). The toxin in its native form is lethal. For chemical inactivation, formaldehyde or aluminum salts are used to neutralize the toxin. This toxin would not generate a strong immune response; therefore, an adjuvant, usually aluminum, is added (72). The vaccine for pertussis may act as an adjuvant for the tetanus vaccine; hence, the vaccine is administered as a combination dose of the diphtheria-pertussis-tetanus (DPT) vaccine.
4.1. Vaccine Delivery Using Adjuvants
An adjuvant is a compound that helps the vaccine enhance the immune response. Although several potential adjuvants have been proposed and used in clinical trials, they are not accepted because of their high toxicity and side effects such as depot development at the site of administration by mineral compounds, oil-based adjuvants, or biodegradable polymers and liposomes (73). Freund’s complete adjuvant (FCA), muramyl dipeptide (MDP), pertussis toxin (PT), and monophosphoryl lipid A (MPL) are the widely used adjuvants, as they act like immuno-stimulators (74).
An ideal adjuvant has minimal side effects with minimized local and systemic reactions, elicits the maximal response with less antigen, and has no carcinogenicity, hypersensitivity, or teratogenicity. It needs to be stable and biodegradable (74). In addition, it should pose physical properties such as high surface area, high pI, and good capacity for adsorption of positively charged proteins (75).
Aluminum compounds such as aluminum hydroxide, aluminum phosphate, and alum-precipitated compounds are the most common adjuvants for human use. Protein antigens such as diphtheria and tetanus toxoids are purified using aluminum phosphate and aluminum hydroxide in the presence of anionic compounds such as bicarbonates and sulfates (76). Aluminum hydroxide shows better adsorption than aluminum phosphate for both TT and DT. Studies prove that serum proteins are adsorbed 10 - 20 times more on aluminum hydroxide, establishing aluminum hydroxide as a superior adjuvant to aluminum phosphate with similar results for FCA (74). Another method of adsorption is to incubate the positively charged gel and the antigen at an optimal pH of 6, followed by stirring continuously overnight (77). Vaccines using calcium phosphate are prepared in a similar manner but a severe Arthus type is observed in the case of TT (78). Recently, the purification of protein antigens is done by using ammonium sulfate and techniques such as ultrafiltration and chromatography.
In a comparative study to check the efficiency of adjuvants for TT purified by ammonium sulphate, ultrafiltration, chromatography, and adsorption onto aluminum phosphate, calcium phosphate, or stearyl tyrosine, it was observed that aluminum phosphate generated the highest titre value of IgG in the first dose, followed by calcium phosphate, while stearyl tyrosine did not yield impressive titre values. However, in the second dosage, the difference in the titre values was negligible between all the three adjuvants. Moreover, TT purified by chromatography showed the highest titre values for all the three adjuvants (79).
Another comparative study demonstrated the efficiency of aluminum hydroxide and calcium phosphate for diphtheria-tetanus (DiT) toxoid. The aluminum hydroxide adjuvant expressed higher titre values for both diphtheria and tetanus toxoid (TT) than the calcium phosphate adjuvant (80).
Liposomes made of dioxyethylene cetyl ether, cholesterol, and oleic acid are non-phospholipid compounds in nature used as adjuvants in TT and DT. Studies show that they elucidate a higher response than FCA aluminum phosphate. TT-encapsulated vaccines sustained more antibody levels than TT vaccines mixed with liposomes. Encapsulated liposomes showed higher anamnestic responses than aluminum phosphate adsorbed to TT (81).
FCA is a water-in-oil emulsion with killed mycobacteria in the aqueous phase. This is one of the most potent adjuvants, as it confers a delayed-type hypersensitivity (DTH) response by directing T lymphocytes to acquire a Th1 pattern (82, 83). FCA causes long-lasting local reactions, ulcers at the site of injection, and toxicity in humans (84). Freund’s incomplete adjuvant (FIA) is a water-in-oil emulsion but lacks mycobacteria. FIA is prepared by using paraffin oil with mannide mono-oleate as a surfactant. It generates immune responses by releasing antigens from oil to stimulate innate immunity (85). The toxicity of FIA is low; hence, it is suitable for human vaccine formulations. Both the adjuvants act by prolonging the life of antigen and stimulating its delivery to the immune system (86).
4.2. Vaccine Delivery Using Micro-Particles
Microspheres exhibit an immunostimulant property, thus acting as antigen delivery vehicles (87). Micro-particles are used as adjuvants for the prolonged and controlled release of toxoid. They reduce the frequency of booster doses and are potent in making the toxoid vaccine a single-dose vaccine (88). Micro-particles are prominently used in the preparation of diphtheria toxoid (DT). The DT is encapsulated in microparticles using the polylactide-co-glycolide polymer based on the solvent evaporation technique (89). Various combinations are made with different sizes of microparticles and release characteristics. These were tested on Sprague rats and the antibody response was monitored and compared with alum immunized control groups for a period of 32 weeks. It was observed that microparticles with single trapped antigen DT had a better response than the combination of DT and TT because of the presence of antigenic competition among both the antigens resulting in poor antigen presentation (90). Microparticles with a single polymer were less effective for long-term antibody formation while the combination of the vaccine and antigens with alum adsorbed to it gave the best response in the formation of antibody titres and the duration of the response (90).
The advantages of microparticles in vaccine development are related to their biodegradable nature, ability to generate a potent immune response, and ability to control the release of antigens by manipulating their composition, molecular weight, and crystallinity of the polymer (91, 92). The variation in size of the microparticle controls the particle uptake by antigen presenting cell (APC). Smaller microparticles (< 10 µ) are taken by macrophages while the bigger ones are not taken by macrophages as these particles exhibit a stronger adjuvant activity than microparticles of greater than 10 µ (91, 93).
The microparticles are prepared by the solvent evaporation method in which the larger microparticles with DT toxoid are diluted and emulsified using a Silverson homogenizer and polymer solution in methylene chloride (94). This emulsion is added to distilled water containing polyvinyl alcohol resulting in the formation of a water-oil-water (w/o/w) emulsion. The microparticles are obtained after evaporating the w/o/w emulsion. Smaller microparticles can be prepared by varying the polymer concentration and stirring speed (94).
Biodegradable microspheres are a type of microparticles made of poly(L-lactic acid) (PLA) or poly(D, L-lactic/glycolic acid) (PLGA) (95). A biodegradable microsphere for tetanus toxoid is prepared by the solvent extraction or solvent evaporation method in multiple emulsion systems (94, 96, 97). The protein was analyzed at different physical conditions to check for its antigenicity and integrity. A partial loss in antigenicity was observed because of lyophilization and the nature of the organic solvent (98). Varied sizes (ranging from 3 kDa to130 kD) of PLA and PLGA showed good protein loading efficiency (99). Protein release is influenced by polymer weight and composition. A study demonstrated that large microspheres were degraded slowly, hence having a low titer value while smaller microparticles exhibited a burst and continuously increasing release rate (99). PLA and PLGA showed a constant release pattern and the release rate of PLA was lower than that of PLGA (96). The microencapsulated vaccines are more immunogenic than the fluid vaccines as determined by the IgG levels (95) although the duration and response of antibody did not much differ (100).
Microspheres may be used to carry antigens at the targeted delivery sites; for example, they transfer the TT vaccine in mucous associated lymphoid tissue (MALT) (101). The microspheres are made of polystyrene, poly(methyl methacrylate), poly(hydroxybutyrate), poly(DL-lactide), and poly(lactide) with varying ratios of lactide to glycolide. When the drug was orally administered, it showed good absorption in the payer’s patches in the small intestine. The microspheres coated with ethyl cellulose, acetate hydrogen phthalate, or cellulose triacetate showed very low uptake. The tissue penetration is specific and limited to the diameter of 10 µ. However, microspheres with diameters of smaller than 5 µ are transported through the efferent lymphatics within the macrophages while the ones with a greater diameter remain fixed at the payer’s patch (102). The effective delivery of microspheres in MALT is associated with their ability to produce secretory IgA. Microspheres are prepared using endotoxin or coumarin-6 dye by microencapsulation procedures. It was observed that hydrophobic particles were readily phagocytized by the reticular-endothelial system (102).
5. Conjugate Vaccine
A conjugate vaccine is a covalent vaccine made by joining a weak antigen to a strong antigen, resulting in the increased immunogenicity of the weak antigen. The weak antigen is usually a polysaccharide that is attached to a strong protein antigen. Recently, peptide-protein or protein-protein conjugates have also been developed.
The response of B cells to the capsular polysaccharide is T cell-independent, implying that B cells can produce antibodies without the help of T cells. Normally, polysaccharides cannot be loaded by themselves on MHCs of the antigen presenting cells (APCs) because MHCs bind only to peptides; therefore, a T cell response can be induced by conjugating a polysaccharide (103). The target polysaccharide antigen is linked to the carrier peptide that is made available to bind to the MHCs molecule for activating T cells. T cells generate a vigorous immune response and a long-lasting immune memory; hence, the conjugate vaccine is very effective in the prevention of invasive bacterial diseases (104).
5.1. Conjugation by Covalent Linkage to Carrier Proteins
A polysaccharide conjugate vaccine consists of a polysaccharide that is covalently attached to the carrier protein to provide epitopes for T cell-independent antigens. The polysaccharide of known molecular size is chemically purified for the generation of chemically reactive groups that can form a bond with the carrier protein. The methods used for polysaccharide activation are the periodate oxidation of vicinal hydroxyls and cyanationof hydroxyls (105). The size of the polysaccharide may be noted after purification as low-molecular size impurities result in inefficient conjugation.
The factors considered for conjugation include the polysaccharide to protein ratio and the percentage of non-conjugate saccharide. Yield and conjugate stability play determining roles, as well. Usually, less than 20% of the activated polysaccharide become conjugated, which can be increased by improving the conjugation methods through the generation of highly reactive groups (105).
Carrier proteins bind to the MHCs of APC. The carrier proteins used in conjugate vaccine preparation are genetically modified cross reacting material (CRM) of diphtheria toxin, tetanus toxoid (TT), meningococcal outer membrane protein complex (OMPC), DT, and hemophilus influenza protein D (HiD) (103). These carrier proteins are effective in increasing the vaccine immunogenicity but they vary in quantity and avidity of the antibody they carry.
CRM197 is a nontoxic variant of diphtheria toxin. A point mutation by glycine substituting glutamic acid at position 52 of the polypeptide sequence results in the elimination of enzyme activity and toxicity. CRM 197 has more lysyl side chains available for conjugation and does not require inactivation by formaldehyde. It requires formaldehyde detoxification and is obtained at about 100% purity. The size is about 63 kD (106). For example, the meningococcal serogroup C vaccine was developed by conjugating it with CRM197 (107).
DT is derived from Corynebacterium diphtheriae, detoxified by formaldehyde, and purified from ammonium sulfate fractionation and distillation. The size is about 63 kD (108). A tetravalent meningococcal vaccine of serogroups A, C, Y, and W135 conjugated to diphtheria toxin is such an example (107).
TT is isolated from Clostridium tetani by detoxification with formalin, purification with ammonium sulfate, and filter sterilization before use. It is about 140 kD in size (109). The meningococcal serogroup C vaccine can also be conjugated to TT as first introduced in the UK (107, 110, 111). The meningococcal serogroup A vaccine is produced using TT as a carrier protein (112) and it was observed to be efficient in providing immunization to toddlers and school children (113).
OMPC is isolated from the N. meningitidis serogroup B strain 11 outer membrane protein complex. It is purified by detergent extraction, ultracentrifugation, defiltration, and sterile filtration. The size is about 37 kD (114).
HiD is an antigenically conserved surface lipoprotein isolated by solubilization with sonication and sarkosyl-extraction by SDS-PAGE. It is used in a non-acylated active form. The size is 42 kD (115).
HiB polyribosylribitol phosphate (PRP) conjugate vaccines show local reactions such as redness, pain, and swelling in infants. It was observed that the inflammatory reactions were less frequent in children who received the vaccine with carrier proteins D and CRM than those receiving the vaccine with carrier proteins OMP and T after three doses of the vaccine (104). The first injection showed a high rate of irritability, crying, and fever in T carrier proteins but not in subsequent injections. HiB-OMP showed lymphadenopathy, hypersensitivity, abscess, and febrile seizures (103).
The vaccine stability changes with the molecular size of polysaccharides and the percentage of free polysaccharides. It has been reported that saccharides of shorter chain lengths are better to develop T cell-dependent antibody responses (116). Oligosaccharide-T with an average length of 14.5 kDa is a superior immunogen to that with the average long chain of 27 kDa. However, the presence of conformational epitopes is an important determinant of the optimal length of the oligosaccharide used in the conjugated vaccine (116).
The pneumococcal conjugate vaccine is composed of a bacterial polysaccharide conjugated with a carrier protein. There are several valency variants of this vaccine. In a placebo-controlled trial in adults aged more than 65 years, the efficacy of 13-valent polysaccharide conjugate vaccine (PCV13) was determined (117) for vaccine type-community acquired pneumonia and vaccine type-invasive pneumococcal disease (IPD) (118). It was observed that PCV13 had significant efficacy in the prevention of pneumococcal disease in adults over 65 years of age (119). Another study demonstrated that the non-conjugated pneumococcal vaccine is not effective in preventing pneumonia (120). The converse was demonstrated in the case of PCV13 versus the 23-valent pneumococcal polysaccharide vaccine (PPSV23) (121).
6. Conclusions
LAVs, inactivated vaccines, toxoid vaccines, and conjugate vaccines have been available since the 20th century, thus referred to as traditional vaccines. Traditional vaccines have found application in immunization against a number of infectious diseases over the years. These vaccines have also been improved and improvised to increase their efficacy while reducing the dosage and toxicity. In the 21st century, many advancements have occurred in vaccination, e.g., the development of recombinant DNA vaccines that provide a long-term immunity similar to LAVs, but do not contain attenuated organisms similar to inactivated vaccines; however, rDNA vaccines are still under preclinical and clinical trials for a majority of diseases. Another development in modern vaccinology is edible vaccines; however, the preclinical trial results are not as promising as rDNA vaccines. Nanotechnology has been developed to produce antigen-carrying vehicles. However, the biocompatibility of nanoparticles and the rate of antigen release from nanoparticles are the limiting factors in this development.
