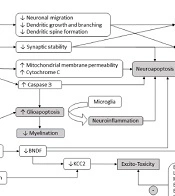


1. Context
Every year, millions of children undergo exposure to general anesthesia for various surgical and diagnostic procedures (1, 2). Preclinical studies reveal that both GABA agonists and NMDA receptor antagonists can induce morphological and functional changes in several neuronal transmission systems, alter dendritic growth and brain connectivity, induce neuroapoptosis (3, 4), and produce other neuronal alterations that can persist over time and be transmitted intergenerationally to the offspring, as will be explained later (5) (Figure 1). These effects are related to age, dose, number of anesthetics received, and duration of exposure to said drugs; they can be reversible or permanent and caused by direct or indirect perturbations on neurons.
, NMDA (N- methyl -D-aspartate), C-fos (cellular Finkel-Biskis-Jinkis murine sarcoma virus osteosarcoma oncogene), IP3 (Inositol 1,4,5-triphosphate), KCC2 (potassium chloride transporter), BCL-XL (B-cell lymphoma-extra-large), Tet1 (Tet methylcytosine dioxygenase 1), H3 (histone 3). + expresses an agonistic effect; - expresses an inhibitory effect.")
Schematic representation of the main cellular mechanisms involved in neurodevelopmental disorders associated with general anesthesia in children. GABA (gamma-aminobutyric acid), NMDA (N- methyl -D-aspartate), C-fos (cellular Finkel-Biskis-Jinkis murine sarcoma virus osteosarcoma oncogene), IP3 (Inositol 1,4,5-triphosphate), KCC2 (potassium chloride transporter), BCL-XL (B-cell lymphoma-extra-large), Tet1 (Tet methylcytosine dioxygenase 1), H3 (histone 3). + expresses an agonistic effect; - expresses an inhibitory effect.
Recent systematic reviews and meta-analyses have reported neurodevelopmental disturbances in humans exposed to general anesthesia during the early stages of neurodevelopment (6, 7). Behavioral problems have been identified after exposure to a single anesthetic in school children (6), compromises in different neurodevelopmental domains with prolonged or repeated exposure to anesthetics (8), and regardless of the type of exposure (single or multiple), increased incidence of behavioral problems, worse academic performance, and scores in nonverbal reasoning, general development, executive function, language, cognition, and motor function (7).
These findings have turned scientific attention to the consequences that general anesthesia can have in the early stages of human life. This manuscript aims to review the cellular mechanisms involved in neurodevelopmental disorders and neurotoxicity caused by general anesthesia.
2. Development
Animal and in vitro studies demonstrate worrying morphological and functional alterations in young neurons and glia induced by different anesthetic drugs. General anesthetics produce significant apoptotic damage, synaptic dysfunction, alterations in brain connectivity, and defective formation of neuronal circuits. They also activate other mechanisms of neuronal death, including mitochondrial dysfunction, excitotoxicity, dysregulation of BNDF, abnormal cell cycle reentry, and interference with the cytoskeleton assembly (3). The suppression of neuronal traffic and sensory information caused during general anesthesia affects the trophism necessary for neurogenesis and context-dependent modulation of neuroplasticity. The most important cellular mechanisms involved in the genesis of these alterations are described below.
3. Neuroapoptosis
Neuroapoptosis is a normal and crucial process for constructing functional brain connectivity. It primarily occurs between the 24th week of gestation and the 4th week of extrauterine life but can extend up to the first year. Programmed cell death affects all neuronal populations and is vital for properly forming neural circuits that define sensory, motor, or cognitive functions.
Developing neurons possess active apoptotic machinery designed to eliminate neurons that have migrated to abnormal locations, display misplaced axons during their pathfinding, or have established connections with inappropriate targets related to their afferent inputs (9). During neurodevelopment, neurons are overproduced to compete for contact with partner cells and ensure adequate innervation of their targets. This surplus also applies to glia, which provides myelination to axons, as discussed below. Glutamate and GABA facilitate organized neuroapoptosis. The action of GABAergic anesthetics and NMDA blockers induces synaptic deprivation, reduces dendrite formation, disrupts neuronal migration, and deems many neurons redundant by altering the homeostatic and cellular signaling environment and triggers their apoptotic mechanisms (9). These alterations are manifiest in later developmental stages as behavioral and cognitive dysfunctions (10).
General anesthetics induce significant and prolonged alterations in the morphogenesis, fission, and fusion of mitochondria, affecting the ATP source necessary for synthesizing, transporting, releasing, and reuptake of neurotransmitters during synaptogenesis, consequently causing neuronal apoptosis. Neuroapoptosis is triggered by damage to both mitochondria and the endoplasmic reticulum. This process is initiated by the decrease in expression of antiapoptotic proteins from the Bcl-2 family, such as Bcl-xL. This reduction leads to increased permeability of both the endoplasmic reticulum and mitochondrial membranes, resulting in the release of cytochrome C and the activation of caspases (11). In immature rats, isoflurane stimulates the release of Ca2+ from the endoplasmic reticulum (ER) by activating inositol 1,4,5-trisphosphate (IP3) receptors. This activation, in turn, stimulates the mitochondrial protein Bcl-xL, subsequently inducing apoptotic neuronal death in the brain. This underscores the critical role that intracellular calcium and the ER play in synaptic transmission and neuronal survival. Neurons that survive may exhibit functional alterations, as observed in the hippocampus of rats, an area crucial for learning and memory.
Glioapoptosis induced by Isoflurane has been observed, particularly in oligodendrocytes, which are numerous in the white matter during the neonatal period, actively myelinating, but rare in the cerebral gray matter. Since a single oligodendrocyte myelinates multiple axons, glioapoptosis can locally impair multiple axons' myelination and nerve conduction (12). These deceased cells are subsequently phagocytosed by microglia, leading to neuroinflammation and further hindering oligodendrocyte maturation and myelination in neonatal white matter. It is noteworthy that isoflurane-induced oligoapoptosis is mediated by Caspase 3, unlike oligodendrocyte death associated with hypoxia and perinatal ischemia, primarily involving the initial loss of pre-myelinating oligodendrocyte progenitors through an excitotoxic (necrotic) mechanism (12).
The histological gold standard for assessing neurotoxicity is activated Caspase 3 (AC3), a well-validated and robust biomarker of apoptosis. Immunohistochemistry targeting AC3 reveals two fundamental aspects of the neurotoxic impact of general anesthetics: their rapidity and generalized distribution. In neonatal rodents subjected to general anesthesia for 6 hours, neuroapoptosis began shortly after its administration; AC3-immunoreactive neurons were detected at 2 hours in the subiculum, caudate nucleus, and putamen, and at 6 hours in the neocortex, hippocampus, superior and inferior colliculi, and cerebellum. However, with prolonged anesthesia, neuroapoptosis extends to more profound subcortical structures, including the thalamus (13). In other words, it is a widespread phenomenon. Non-human primates also display similar patterns of AC3 immunoreactivity to those seen in rodents but additionally undergo oligodendrocyte apoptosis (12), suggesting that aberrant myelination may be a uniquely higher-order mammalian neuropathological feature of neonatal anesthesia (5).
4. Excitotoxicity
Excitotoxicity is a neuronal injury process induced by increased intracellular calcium concentrations in response to overstimulation of glutamate receptors, such as the NMDA receptor and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA) (14). Neuronal depolarization activates NMDA receptors and voltage-gated calcium channels that contribute, through the increase in intracellular calcium, to shaping neuronal activity and establishing adult neuronal circuits, through the formation of new synapses and elimination of others.
The neuronal demise triggered by the NMDA receptor antagonist ketamine results from the compensatory upregulation of NMDA receptors, resulting in calcium-mediated neuronal toxic effects, elevated generation of reactive oxygen species, and ultimately, cell death (15).
Another plausible alternative is that the cognitive alterations associated with exposure to anesthesia in the neonatal period, observed in a wide range of laboratory animals, are not due to neuroapoptosis as an initial step. Instead, it could arise from more nuanced yet enduring neuronal structure and synaptic communication alterations. This occurs during periods when rapidly developing and consequently susceptible neurons are subjected to non-physiological conditions induced by general anesthetics, giving rise to a form of chronic neuropathy (3).
5. Effects on Chloride Cotransporters
GABA, the main inhibitory neurotransmitter in the adult brain, plays a major role in the early assembly and formation of neural circuits during neural development. Unlike what happens in the adult brain, GABA is the main excitatory neurotransmitter during embryonic and early postnatal development due to the expression of the chloride importer Na+-K+-Cl− cotransporter (NKCC1) (16). Because NKCC1 maintains a high level of intracellular chloride, activation of the GABAA receptor promotes the release of chloride to the outside of the neuron and, therefore, neuronal depolarization (excitatory effect). This GABA-mediated excitation of immature neurons is central to several ontogenetic processes in the perinatal period. This process continues to be active during the initial months of life in human infants before transitioning to its neuroinhibitory function (17).
Sevoflurane induces downregulation of the cation-chloride cotransporters NKCC1 and KCC2 in the brain of neonatal mice (16) through genomic and epigenomic effects on Nkcc1 and Kcc2 gene expression (18). Indeed, in vivo, anesthetics significantly decrease BDNF protein and mRNA levels (19). Considering that BDNF protein plays a regulatory role in the expression of the Kcc2 gene during neurodevelopment and epigenetic changes impact the Nkcc1 and Kcc2 genes, sevoflurane can directly downregulate KCC2 by altering the acetylation or methylation status of the Slc12A5 gene for KCC2. Additionally, it can indirectly reduce neuronal levels of the BDNF protein (16). Thus, exposure to sevoflurane during the neonatal period can induce a more immature state in neurons, leading to GABA depolarization and favoring neuronal hyperexcitability, resulting in an excitotoxic state.
On the other hand, Bumetanide has been found to decrease the excitotoxic response of neurons due to its blocking effect on the NKCC1 channel (20). Interestingly, other drugs with neuroprotective properties, such as lithium, melatonin, estrogen, erythropoietin, estradiol, and dexmedetomidine, have demonstrated protective effects in animals against the neurotoxic effect of general anesthetics (21).
6. Effects on Synaptic Plasticity and Neurotransmission
Synaptic plasticity refers to the intricate process through which neurons manage, encode, and retain information. This dynamic mechanism depends on the synaptic activity of the neurons. Achieving this involves fine-tuning synaptic morphology by regulating intracellular signaling pathways that impact gene transcription. The architecture of synapses is subsequently altered by incorporating or excluding neurotransmitter receptors, neurotrophic factors, and scaffolding proteins. Consequently, this modification affects how neurons respond to synaptic neurotransmission, ultimately governing their overall function. These genetic, chemical, and morphological mechanisms are perfectly coordinated to guarantee the faithful recording of molecular memory.
Anesthetics used during critical developmental stages perturb neural biochemistry essential for synaptic neurotransmission, gene morphology, and expression, as well as the plasticity of neural circuitry, consistent with the development of cognitive and behavioral deficits (22).
7. Effects on Synaptic Morphology
As previously stated, during the neonatal period, general anesthesia induces alterations in neurotransmission coupled with disruptions in synaptic architecture and neuronal morphology. For instance, the dendritic arborizations of pyramidal neurons exhibit dense coverage with dendritic spines. These bulbous protrusions capture excitatory information from the terminals of presynaptic axons. At the subcellular level, the neuropil is empty and disorganized; there is a decrease in presynaptic axon terminals and in the number of mitochondria in the synaptic terminals, leaving neurons in a low-energy state that affects synaptic plasticity, a phenomenon that has been found in non-human primate neonates (23). These changes explain why general anesthesia disrupts information processing in immature brains.
Since several epigenetically regulated genes modulate synaptic plasticity, neonatal anesthesia can be expected to have deleterious effects on the genome and epigenome.
8. Effects on the Genome
There is evidence of the alteration caused by anesthetics in dozens of genes, most related to the essential and survival functions of the neuron, especially the gene for BDNF and the c-fos oncogene, both critical for neuronal function.
The Bdnf gene is essential for neurological development and regulates many aspects of synaptic plasticity and neural circuit function throughout life. Its gene expression is upregulated in response to neuronal stimulation. It also modulates synaptic neurotransmission by increasing the number of presynaptic vesicles and neurotransmitter release while affecting the postsynaptic receptor kinetics of both glutamate and GABA (24). The Bdnf gene plays a role in these processes through the interaction of the BDNF protein with tropomyosin-related kinase B (TRKB) receptors, contributing to memory, learning, and synaptic stability (25, 26).
Conversely, when BDNF binds to the pan-neurotrophin 75 receptor (P75NTR), it can remove inactive or dysfunctional synapses, promote apoptosis, and potentially impair synaptic plasticity. On the other hand, BDNF binding to the pan neurotrophin 75 receptor (P75 NTR) can promote apoptosis, eliminate inactive or dysfunctional synapses, and compromise synaptic plasticity. General anesthesia with GABAergic drugs and glutamate antagonists markedly reduces the BDNF levels in neonatal hippocampus and thalamus extracts in rodents and non-human primates (19, 27, 28). However, it appears that the downregulation of BDNF caused by general anesthesia does not occur in the cerebral cortex, being a compensatory mechanism during crucial moments of neurological development that allows maintaining the homeostasis of BDNF signaling, which is essential during synaptogenesis and synaptic restructuring (5).
The neuronal activity also controls the expression of the early gene c-fos, whose protein product couples extracellular signal transduction to the transcriptional machinery of target genes. During the neonatal period, general anesthetics significantly downregulate C-FOS (Cellular Finkel-Biskis-Jinkis murine sarcoma virus osteosarcoma oncogene) protein expression in the cortex, thalamus, and hippocampus (19, 27).
Changes in the expression of genes that regulate early developmental responses to neurotransmitters have also been reported, such as the already described cation-chlorine cotransporters NKCC1 and KCC2 (16), and the decrease in the expression of the Reelin gene (29), important for the control of dendritic growth, branching, spine formation, neuronal migration, synaptic plasticity, and synaptogenesis (30).
Dexmedetomidine is a type of α2 adrenergic receptor agonist that is widely used as an adjunct to general anesthesia. Dexmedetomidine has been shown to inhibit general anesthesia-induced hypoxia/reoxygenation-induced neuroapoptosis in rats by blocking the activity of gap junctions through diminishing Cx32 protein levels (31). In anesthetized animals, Dexmedetomidine's antioxidant, anti-inflammatory, and anti-apoptotic properties have shown neuroprotective effects, attenuating the cognitive impairment induced by isoflurane, which was evidenced by a higher expression of BDNF (32).
9. Effects on the Epigenome
Epigenetics studies DNA changes that do not involve alterations in the underlying sequence and are produced by certain biochemical processes that cause remodeling of the chromatin structure, ultimately changing the level at which genes are activated and deactivated. The arrangement of chromatin involves its packaging into nucleosomes, consisting of a short DNA strand wound around octamers of histones H2A, H2B, H3, and H4 proteins. Histones possess extended amino-terminal tails that are susceptible to modifications like ubiquitination, phosphorylation, acetylation, and methylation. Condensed chromatin restricts access to the genetic transcription machinery, whereas relaxed chromatin enables reading and transcription. Among the mechanisms that regulate chromatin structure, DNA methylation and histone acetylation seem to play an important role in the neurotoxicity produced by general anesthetics.
DNA methylation is the chemical process whereby a methyl group (-CH3) is added to a cytosine base at the carbon 5 position (5 mC methylation) by DNA methyltransferases (Dnmt), protecting gene promoters from the transcriptional machinery and generally silencing gene transcription. Conversely, histone acetylation relaxes the chromatin structure, allowing gene transcription. Histone acetylation is facilitated by histone acetyltransferase (HAT) and deacetylation, which leads to chromatin condensation and restricted gene transcription by histone deacetyltransferase (HDAC). The inhibition of HDAC leads to elevated histone acetylation, resulting in increased expression of the c-fos and Bdnf genes. This, in turn, fosters the formation of new memories (19). Different studies show that neonatal anesthesia can change chromatin structure towards a more repressive transcriptional state through DNA hypermethylation and histone deacetylation (18, 33).
When examining the histone acetylation status in the hippocampus of neonatal rats, it is observed that acetylated H3 and H4 levels are markedly reduced in the long term, corresponding to a neuropathological consequence of exposure to neonatal anesthesia (5). Similar effects occur with cyclic adenosine monophosphate response element-binding protein (CREB), a cellular transcription factor responsible for regulating many target genes crucial for acquiring and retaining new memories (19). In turn, reducing H3 and H4 acetylation and CREB leads to negatively regulated transcription of the target genes c-fos and Bdnf, which play a crucial role in neuronal development (34).
Repeated exposure to sevoflurane in newborn rats increased Dnmt1 and Dnmt3a, decreased Tet1 (an enzyme that performs DNA hydroxymethylation in oligodendrocytes necessary for myelin repair), and promoted hypermethylation of the Shank2, Psd95, Syn1, and Syp genes. Subsequently, this downregulated the expression of synaptic genes and ultimately caused impairment of cognitive, social, and spatial memory later in life (5).
Another interesting aspect is that alterations in the epigenomic control of gene expression can be transmitted from parents who underwent anesthesia during their neonatal period, transferring these changes to their offspring, even if the offspring themselves had no prior exposure (35). Thus, mothers who received general anesthesia with sevoflurane before pregnancy revealed a reduction in the expression of the Kcc2 gene. In the offspring, there were substantial increases in Kcc2 gene methylation, destruction of the neuropil and mitochondria, substantial dysregulation of synaptic transmission, and, in addition, a moderate decrease in hippocampal-dependent learning and memory. Reduced expression of the Kcc2 gene was observed in mothers exposed to sevoflurane before pregnancy. This gene has been linked to the development of autism spectrum disorders in the human population (13).
The present manuscript focuses on the cellular mechanisms involved in the neurodevelopmental alterations caused by general anesthesia based on data from experiments on animals in laboratory settings, so its extrapolation to humans must be approached with caution.
10. Conclusions
Preclinical studies show evidence of various cellular mechanisms associated with neurodevelopmental abnormalities in children exposed to GABAergic anesthetic drugs and NMDA antagonists. These include mechanisms that involve the cellular energy machinery, synaptic neurotransmission, and neuronal morphology through the alteration of different cell signaling and transmission proteins, ultimately leading to neuro- and gliopathy, excitotoxicity, myelination, neuronal plasticity alteration, and neuroinflammation. In addition, general anesthetics cause the activation of epigenetic mechanisms that affect gene expression and explain the persistence of these alterations over time. Some medications, such as dexmedetomidine, may have a protective role against these disorders.
