


1. Introduction
Malignant peripheral nerve sheath tumor (MPNST) is a type of connective tissue cancer. Due to its origin and behavior, this tumor is classified as a sarcoma. More than half of patients have neurofibromatosis as an underlying disease (1). Symptoms include painless limb edema, decreased limb movement, pain, and neurological symptoms include numbness and dizziness (1). MPNST is probably a genetic disease because it is found in families, but it is not yet clear (2). Treatments of neurofibrosarcoma are similar to other cancers, and surgery is an option. Removal of the tumor and surrounding tissues can be critical to patient survival. For separate, localized tumors, surgery is often followed by radiation therapy to the removed area to reduce the chance of recurrence is needed (3).
2. Case Presentation
A 4-year-old boy who was normal in terms of growth and development presented with limitation of hand movements with sweating and numbness of the right forearm that lasted for about 2 months. The patient has had various visits to pediatricians, orthopaedists, and neurologists and was subjected to undergone various examinations, including electromyography (EMG), nerve conduction velocity (NCV), whole-body bone scan, forearm X-ray, and various laboratory tests, including CBC, ESR, LDH and alkaline phosphatase that all have been normal. Finally, the patient was referred to a pediatric hematologist-oncologist, and after performing a chest X-ray, a soft tissue mass was seen in the right lung apex (Figure 1).
")
Chest X-ray shows a soft tissue mass in the apex of the right lung (arrow)
Regarding family history, his mother had a brain glioma, which was resected four years ago without any further complication.
On examination, he had decreased muscle forces. Anesthesia and reduction of limb movements were the most common complaints. All other examinations were normal. No evidence of neurofibromatosis was seen in the patient.
A cervical magnetic resonance imaging (MRI) and chest CT-scan of the cervical spine revealed the evidence of an enhancing mass lesion at the right para-tracheal region adjacent to T1, T2, and T3 levels with extension to the right side of the neural foramen, measuring 37 × 20 mm in size with extension to the mediastinum (Figure 2). Also, the MRI of the brain was normal.
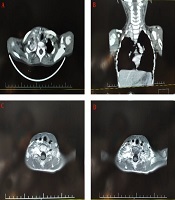
 scan and magnetic resonance imaging (MRI). A laminar heterogeneous mass with a necrotic center of approximately 32 × 40 mm is seen in the right paravertebral region (T1-weighted and T2-weighted scans). No pulmonary parenchymal invasion was observed, and no bone lesions were seen; A, axial view of contrasted CT-scan; B, coronal view of contrast CT-scan; C and D, axial view of T1-weighted MRI contrast.")
Cervical computed tomography (CT) scan and magnetic resonance imaging (MRI). A laminar heterogeneous mass with a necrotic center of approximately 32 × 40 mm is seen in the right paravertebral region (T1-weighted and T2-weighted scans). No pulmonary parenchymal invasion was observed, and no bone lesions were seen; A, axial view of contrasted CT-scan; B, coronal view of contrast CT-scan; C and D, axial view of T1-weighted MRI contrast.
Histopathological sections of tumor cell sheets and fascicles of elongated to spindle-shaped cells showed a moderate degree of pleomorphism and atypia. Immunohistochemistry (IHC) results were positive for CD34, Ki67, MIC2, S-100, and Vimentin and indicated high levels of them. VMA and HVA of 24-hour urine were collected and were normal.
Based on the history, physical examination, laboratory findings, plus imaging, the final diagnosis of paravertebral region MPNST was made for the patient by biopsy and approved by IHC. The tumor was near the resection margins, and lymph nodes were free of the tumor. The metastatic evaluation was normal, as well. He underwent chemotherapy for six months after the operation. After surgery, the patient was discharged and followed up by an oncologist and radiation oncologist. The patient was in a stable condition for six months and is currently in good condition.
3. Discussion
MPNST or malignant neurilemmoma is known as malignant schwannoma or neurogenic sarcoma and is a connective tissue cancer around the nerves. It is classified in the sarcoma group due to its origin and behavior (1, 4). About half of the patients diagnosed are patients with neurofibromatosis. The risk of developing MPNST in patients with type 1 neurofibromatosis is 8% to 13% (1). MPNST etiopathogenesis is believed to be associated with neurofibromatosis with loss of the 17q chromosome arm sequence, including complete inactivation of the neurofibromatosis-1 (NF-1) gene (2).
MPNST accounts for about 5% - 10% of soft tissue sarcomas, of which only about 8% - 16% occur in the head and neck (3). It is usually can be found in the lower extremities, retroperitoneum, trunk, upper extremities, head, and neck (1). In the head and neck regions, frequent sites are the nasopharynx, paranasal sinus, nasal cavity, oral cavity, orbit, cranial nerves, larynx, parapharyngeal or pterygomaxillary space, minor salivary glands, and the thyroid gland (1). Barnes et al. showed a case of an extremely rare presentation of this malignancy owing to the involvement of the paranasal sinus and nasal cavity (5). MPNSTs generally present as a painless, enlarging mass with associated numbness throughout the length of the affected nerve. They mainly affect the 20 - 50-year-old age group and have an equal gender distribution (4, 6).
Imaging studies show the location and spread of disease and metastasis. Fine needle aspiration and biopsy are used for diagnosis. Histologically, these tumors do not have a distinctive and classical appearance. Typical findings are the presence of high-mitotic spindle cells and indeterminate cytoplasmic boundaries arranged in bundles or branches. IHC plays an important role in the diagnosis and elimination of fibrosarcoma, synovial sarcoma, and fiber histiocytoma. MPNST is particularly positive for the S-100 protein (7-10).
S-100, Leu-7, and myelin basic protein can be used to detect nerve sheath differentiation, and they are immunoreactive for vimentin and not for HMB-45. S-100 immunoreactivity is focal and spread in 50% - 90% of MPNSTs (7, 11). The same marker was used in the present case, which showed scattered tumor cells staining positive and confirming the diagnosis.
Treatment for this tumor is mainly surgery, and the goal is to completely remove the tumor with sufficient margins, which has the best results. Radiotherapy can be used before, during, and after surgery. Radiotherapy has also been very effective in reducing recurrence (12).
3.1. Conclusions
We presented an extremely rare case of MPNST in a 4-year-old boy.
