



1. Background
The decreased effect of β-lactam antibiotics on Gram-negative pathogens is a growing clinical problem, leading to antibacterial drug resistance in organisms such as Stenotrophomonas maltophilia, Pseudomonas aeruginosa, Klebsiella pneumoniae, and Escherichia coli (1, 2). Each year, about 2.8 million people are diagnosed with bacterial infections in the United States who are resistant to one or two types of antibiotics, leading to 35,000 deaths (3). Bacteria become resistant to antibiotics by mutations, and bacterial resistance manifests a vital need for novel antimicrobial agents (4, 5). Most bacterial resistance is due to the overexpression of β-lactamases, which can hydrolyze and inactivate many β-lactam antibiotics (6, 7). Antibiotic resistance has increased the attention of the World Health Organization in recent years as an important challenge for humans (7). Although β-lactam antibiotics such as carbapenem, cephalosporins, monobactams, and penicillins are currently used, antibiotic resistance necessitates studies to find new compounds with better and broader activity (8-10).
In the last two decades, Metallo-β-lactamases (MBLs) have been identified as major role-players in the mechanism of resistance to carbapenems, which are the last resort of β-lactam antibiotics for the treatment of infections caused by multi-drug-resistant pathogens (10, 11). β-lactamases (BLs) are arranged in two classes, including MBLs (metallo-BLs) containing a hydroxyl ion (as active nucleophile) and one or two zinc ions (in the binding site), and SBLs (serine-BLs) utilizing a catalytic serine for antibiotic hydrolysis (12, 13). MBLs are zinc-dependent enzymes that hydrolyze almost all β-lactams, such as cephalosporins and carbapenems (14). SBLs have Ambler classes (A, C, and D), but MBLs cover class B with three subgroups (B1, B2, and B3). Also, chromosomal and plasmid-mediated genes can encode BLs (7).
The unique structure of the active site, di-zinc coordination, and water bridge hydrolysis mechanism enable subclass B1 to inactivate a variety of substrates (15). The β-lactam ring of antibiotics can be hydrolyzed by SBLs based on the serine residue through an enzyme-acyl intermediate around the active site. On the other hand, MBLs can catalyze the hydrolysis of the β-lactam ring by utilizing the Zn ions (one or two) in the active site (16, 17). The most common clinical problems in MBLs are with the B1 subclass, including VIM (Verona integrin-encoded Metallo-β-lactamase), NDM (New Delhi Metallo-β-lactamase), and IMP (IMiPenamase) (18, 19). VIM is currently the most common among the B1 subclass of MBLs in P. aeruginosa, and the primary source of global prevalence is VIM-2 (20). Developmental studies have critical roles in finding new inhibitors to overcome MBLs and treat bacterial resistance (21). In parts of Asia, VIM exists in more than 99% of patients with MBL-positive multidrug-resistant strains (22).
Captopril ((D-3-mercapto-2-methylpropranoyl-L-proline) found by Bristol-Myers in Boston) has been used for several decades as a suitable drug for high blood pressure prevention. Captopril also can inhibit different MBLs of all subclasses (23, 24). In the present study, captopril was selected as a positive control for the inhibition of VIM-2 MBL. In addition, virtual screening was chosen to find strong and new inhibitors for time and cost-saving purposes. Also, molecular dynamics simulations were performed by the GROMACS package.
2. Objectives
The purpose of this investigation was to recognize a potent inhibitor for VIM-2 MBL and compare it with captopril as a reference inhibitor.
3. Methods
3.1. Molecular Docking
Natural compounds from the ZINC databank (25) in mol2 format containing 61953 structures were achieved. The VIM-2 MBL crystal structure (ID: 4C1E) in the PDB format was obtained from the RCSB (Protein Data Bank) (23). Both proteins and compounds were prepared, and Maestro 12.5 (26) was employed for the docking study. In addition, the ADME (absorption, distribution, metabolism, and excretion) calculation was performed using an online platform (27). Captopril was chosen as the positive control for docking and molecular dynamics (MD) simulation (23).
3.2. Molecular Dynamic
In the next step, two complexes of ligand-protein (MBL-ZINC00517765 and MBL-Captopril) were extracted from the results of the docking study for MD simulation. The ACPYPE server (28) was used to produce the topology parameters file for compounds. GROMACS software (Amber99sb force field) and TIP3P water model with cubic box were applied to simulate the systems (29, 30). The physiological condition (the salt ion environment (NaCl 0.15M)) was fixed, and the charge was neutralized for three systems by adding Na+ or Cl- (9 Na, 11 Na, 10 NA ions to free MBL, MBL-ZINC00517765, MBL-Captopril, respectively) to the solvent (31). The temperature and pressure were determined by employing NPT and NVT ensembles (1 bar with Parrinello-Rahman barostat and 310K using a v-rescale thermostat) (32). After simulation, the radius of gyration (Rg) and principal component analysis (PCA) were employed to investigate the flexibility and motion, and root mean squared fluctuation (RMSF) with root mean square deviation (RMSD) were used to estimate the fluctuations and H-bond (inter-molecular hydrogen bonds). The simulation time for all systems was 120 nanoseconds.
3.3. Binding Free Energy
Molecular mechanics Poisson–Boltzmann surface area (MM-PBSA) is a reliable method for calculating the binding free energy (ΔGbind). In the present study, the GROMACS Tool (g_mmpbsa) was used to study binding free energy. This approach has been used in computational drug design as an appropriate technique for measuring binding free energy (33-35). The following equation expresses the binding free energy:
G of binding = G of complex – (G of protein + G of ligand)
4. Results and Discussion
4.1. Virtual Screening
The docking study by Maestro 12.5 software determined the power of the new compounds based on inhibitory potential in the active site of VIM-2 MBL. The top nine compounds based on docking energy including ZINC00517765, ZINC04023122, ZINC12659408, ZINC12889936, ZINC01531046, ZINC02108368, ZINC72326107, ZINC20113415, and ZINC10475480 are listed in Figure 1. Molecular docking results (docking energy) for top structures were between -12.29 kcal mol-1 and -10.12 kcal mol-1. Docking's study displayed that ZINC00517765 with -12.29 kcal mol-1 was stronger than the positive control (captopril) with -10.12 kcal mol-1 docking energy.

Figure 1.
Docking energies of Captopril and the top nine compounds, as well as 2D structures, ZINC IDs, and molecular weight
4.2. ADME Investigations
One of the major problems with the usefulness of new inhibitors is their safety inadequacy. Drug development usually encounters various unforeseen challenges in different phases. The potency of the top inhibitors was measured in terms of pharmacokinetics, lipophilicity, and drug-likeness impacts. Captopril and the nine compounds including ZINC00517765, ZINC04023122, ZINC12659408, ZINC12889936, ZINC01531046, ZINC02108368, ZINC72326107, ZINC20113415, and ZINC10475480 were analyzed. Table 1 shows that ZINC00517765 was in the acceptable region as a drug in terms of absorption, distribution, metabolism, and excretion. Also, captopril features were similar to those of ZINC00517765.
Table 1.Pharmacokinetics, Lipophilicity, and Drug-likeness Features of the Top Nine Compounds Compared with Captopril
| Molecule Name | GI Absorption | Log Kp (Skin Permeation) cm/s | Lipinski | Bioavailability Score | Synthetic Accessibility | BBB Permeant |
|---|---|---|---|---|---|---|
| ZINC00517765 | High | -6.49 | Yes; 0 violation | 0.56 | 2.78 | No |
| ZINC04023122 | High | -5.83 | Yes; 0 violation | 0.85 | 2.98 | Yes |
| ZINC12659408 | Low | -6.75 | Yes; 0 violation | 0.56 | 5.04 | No |
| ZINC12889936 | Low | -8.50 | Yes; 1 violation: NorO>10 | 0.11 | 4.21 | No |
| ZINC01531046 | High | -6.63 | Yes; 0 violation | 0.85 | 1.47 | No |
| ZINC02108368 | Low | -7.32 | Yes; 0 violation | 0.56 | 3.76 | No |
| ZINC72326107 | High | -6.85 | Yes; 0 violation | 0.56 | 3.07 | No |
| ZINC20113415 | High | -10.29 | Yes; 0 violation | 0.56 | 3.84 | No |
| ZINC10475480 | High | -8.19 | Yes; 0 violation | 0.56 | 2.76 | No |
| Captopril | High | -7.38 | Yes; 0 violation | 0.56 | 2.47 | No |
4.3. Interactions of VIM-2 MBL with ZINC00517765 Before Simulation
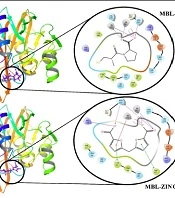
As can be seen in Figure 2, MBL via Asn210 in its active site interacted with captopril by hydrogen bonding. Captopril also interacted with the MBL active site through Zn501 and Zn502. On the other hand, ZINC00517765 formed two hydrogen bonds by interaction with Asn210 from the MBL active site. Furthermore, Tyr67 and Trp87 in the MBL active site formed a pi-pi stacking interaction with ZINC00517765 and Arg205, and Zn501 and Zn502 interacted with the MBL active site, which confirmed that ZINC00517765 was more potent and formed more bonds with the MBL active site.

Figure 2.
Interaction of ZINC00517765 and captopril with the VIM-2 MBL active site before simulation
4.4. Stability Analysis
After MD simulation to evaluate the stability of the crystal structure and changes during the simulation, RMSD (Figure 3A), Rg (Figure 3B), RMSF (Figure 3C), Intermolecular H-bonds (Figure 3D), and PCA (Figure 3E) analyses were performed on MBL-ZINC00517765, MBL-Captopril, and free MBL. The RMSD results for free MBL, MBL-ZINC00517765, and MBL-Captopril were 2.18, 2.13, and 2.09 Å, respectively. The RMSD results showed that from 50 nm to the end of the simulation, all the systems obtained stable conditions, but the stability of the MBL-ZINC00517765 complex was better than those of the other two systems. Based on the Rg analysis, the protein fluctuated during the simulations, and these changes were fewer for the MBL-ZINC00517765 complex, indicating that this complex had better stability. The Rg values for all simulations were between 16.2 and 16.8 Å, but the Rg value for MBL-ZINC00517765 was between 16.2 and 16.7 Å. In addition, RMSF analysis was performed using the simulation results, which showed that free MBL in the residues Val36 with 2.21 Å, Glu38 with 2.20 Å, Asp63 with 3.62 Å, Tyr67 with 2.32 Å, Trp87 with 2.45 Å, His116 with 1.77 Å, and Val211 with 3.32 had the highest fluctuations. Also, Val41 with 2.20 Å, Arg127 with 1.73 Å, Arg141 with 2.75 Å, Cys198 with 1.81 Å, and Leu216 with 2.37 Å had the most fluctuations in the MBL-Captopril system. The RMSF analysis demonstrated that MBL-ZINC00517765 in the residues including Arg109 with 1.38 Å, Glu156 with 2.24 Å, Ser159 with 2.41 Å, Ser161 with 2.92 Å, and Glu232 with 1.98 Å had the most fluctuations. Inter-molecular H-bond analysis revealed that during simulation, ZINC00517765 formed more hydrogen bonds with the active site of MBL than did Captopril. The PCA displayed the movements of proteins. The PCA outcomes showed that MBL-ZINC00517765 had less motion than the other two systems and confirmed the results of other analyses, including Rg, intramolecular H-bonds, RMSF, and RMSD.
, Rg (B), RMSF (C), Intermolecular H-bonds (D), and PCA (E) values for MBL-ZINC00517765 (blue), MBL-Captopril (dark red), and free MBL (yellow)")
Figure 3.
RMSD (A), Rg (B), RMSF (C), Intermolecular H-bonds (D), and PCA (E) values for MBL-ZINC00517765 (blue), MBL-Captopril (dark red), and free MBL (yellow)
4.5. Free Binding Energy
After simulation, the binding free energy was computed for the MBL-ZINC00517765 and MBL-Captopril complexes using MM-PBSA software. According to the RMSD chart, the binding free energy was assessed at the end of the simulation (with minimum fluctuations), and the results exhibited that MBL-ZINC00517765 with -72.29 KJ.mol-1 was stronger than MBL-Captopril with -23.39 KJ. mol-1. Also, the values of electrostatic energy and Van der Waals energy were better for MBL-ZINC00517765 than for MBL-Captopril (Table 2).
Table 2.Molecular Energy Values of MBL-ZINC00517765 and MBL-Captopril Complexes
| Energy (KJ.mol-1) | MBL-Captopril | MBL-ZINC00517765 |
|---|---|---|
| ΔESASA | -17.92 ± 0.27 | -20.93 ± 0.58 |
| ΔEsolv | 253.69 ± 15.17 | 275.57 ± 10.53 |
| ΔEelect | -173.77 ± 7.35 | -215.81 ± 8.36 |
| ΔEvdw | -85.39 ± 4.81 | -111.12 ± 8.85 |
| ΔGbinding | -23.39 ± 5.92 | -72.29 ± 10.74 |
5. Conclusions
According to the results of molecular dynamics, virtual screening, and docking studies, ZINC00517765 can inhibit MBL. The results showed that MBL in the presence of ZINC00517765 has stable conditions and is better than Captopril. Finally, further study on ZINC00517765 is recommended to overcome bacterial infections as a suitable inhibitor. We hope that the results of this study will help researchers to find a drug for MBL.