In this study, clinical isolates of E. faecium comprised the HA clade, while isolates from hospital soil environments constituted the CA and Els clades, consistent with common multi-source E. faecium phylogenetic distributions. A significant trend was observed with ARGs and replicons in each group decreasing from the clinical group to the soil and Els groups, suggesting that environmental E. faecium isolates have lower resistance rates to antibiotics and fewer overall virulence genes. This supports the association between the role of ARGs and replicons and the clinical adaptation of E. faecium.
Considering this association, we explored and identified HA clade-related clinicalʹ group
E. faecium in hospital soil environments based on their phenotypical and molecular biological comparative analysis. Aligning with the HA clade, the clinicalʹ group is distinguished from the CA clade by its shared resistance spectrum, almost identical virulence profile with the HA clade, and population structure analysis showed that the ancestral components of HA and CA clades contributed equally to the clinicalʹ group’s genetic composition. Previous research has shown that the predecessor of the current vancomycin-resistant
E. faecium (VREfm) was the HA clade ampicillin-resistant
E. faecium (AREfm), which evolved into VREfm through horizontal gene transfer of the
van gene (
32). The clinicalʹ group
E. faecium identified in this study also exhibited ampicillin resistance, a trait absent in the CA clade but predominant in the HA clade. Therefore, we believe that the clinicalʹ group
E. faecium parallels historical AREfm in characteristics, with the potential of becoming VREfm by acquiring van genes during infection or colonization in clinical settings.
The resistance phenotype results suggest that ampicillin resistance is not the only differentiation. This study identified a set of antibiotic resistances present only in the clinical and clinicalʹ groups. We assume that such resistance is essential for colonization in patients amidst exposure to various antibiotics in clinical settings, conferring an advantage and resilience against elimination during subsequent treatments. Soil strains lack this uniform resistance distribution, highlighting its significance in the evolutionary adaptation from the CA to the HA E. faecium clades. Notably, the distribution of soil-derived strain clusters in the MST indicates cross-regional transmission in hospital soil, with three clusters, including the clinicalʹ group, spanning from the east to the west of the hospital. This proves that soil E. faecium can traverse long distances with carrier assistance and re-establish in new soil, posing potential health risks due to their similarity to HA E. faecium.
Soil is recognized as a reservoir for ARG-carrying pathogens (
33). Likewise, soil-dwelling
E. faecium can accept resistance genes like
tet(M),
erm(B),
erm(Q), and
mef(A) from
Clostridium perfringens, demonstrating that resistance genes in soil can transfer to
Enterococcus species (
34). Their ability to exchange mobile elements with other soil bacteria may enable
E. faecium to acquire resistance phenotypes or other clinically rare traits. In this study, the mobile element IS1216V combined with the resistance gene
aac(6')-aph(2'') in the clinical and clinicalʹ groups may be involved in such processes. Compared to the clinicalʹ and soil groups, the greater diversity of replicons in the clinical group may contribute to or result from its ultimate adaptation to clinical environments. The clinicalʹ group potentially evolves into HA
E. faecium by acquiring HA mobile elements through infection or colonization in clinical settings or soil environments.
Studies indicate that genetic exchange across
E. faecium clades predominantly occurs from clade B (Els clade) or clade A2 (soil clade) to clade A1 (clinical clade) (
10). This explains why the HA clade contains ancestral components from the other two clades, while these two clades lack HA clade components. We acknowledge that the 13-year time interval between clinical and hospital soil isolates may introduce temporal confounders when comparing resistance profiles. However, longitudinal surveillance studies have demonstrated that the resistome of
E. faecium, particularly hospital-adapted strains, remains relatively stable over time. For example, surveillance data from Italy showed minimal fluctuations in resistance rates to key antibiotics such as ampicillin, linezolid, teicoplanin, tigecycline, and vancomycin across multiple years (
35). Similarly, national data from China’s BRICS program (2016 - 2022) (
36-
40) confirmed consistent resistance rates for these agents among clinical
E. faecium isolates. Within our own dataset, we found no significant differences in resistance phenotypes or resistance gene profiles among patient-derived isolates collected between 2010 and 2019. This internal consistency suggests that, at least within the studied hospital setting, the confounding effect of temporal separation is likely minimal.
We also acknowledge that bacterial resistance mechanisms are dynamic, and that CLSI guidelines for antimicrobial susceptibility testing are updated annually to reflect these changes. However, the antibiotics selected in this study — including ampicillin, penicillin, ciprofloxacin, levofloxacin, erythromycin, vancomycin, and teicoplanin — have consistently remained among the primary agents for E. faecium susceptibility testing over the past decade. Consequently, despite the time interval between patient and soil isolate collections, the major resistance phenotypes compared in this study reflect stable and clinically relevant antibiotic targets. Moreover, our analysis focused on well-established resistance traits rather than newly emerging or rare mechanisms, minimizing the confounding impact of evolving resistance patterns on the validity of our comparisons.
5.1. Conclusions
In conclusion, we isolated five transitional strains from the hospital soil environment. Comparative analyses of resistance phenotypes, ARG distribution, and virulence profiles demonstrate that these strains are transitional between HA and CA E. faecium clades, suggesting their potential for clinical infection. Population structure analysis further showed that the HA and CA clades contributed equally to the ancestral components of the clinicalʹ group, making it a continuum bridging the two clades. The emergence of the clinicalʹ group supports the adaptive transformation process between CA and HA clades and demonstrates that hospital soil environments are not as safe as previously thought. Clinically hazardous E. faecium can adapt to and exploit the hospital soil as a reservoir, facilitating its spread over long distances within hospital environments.






![A sketch map of sampling soil sites within the hospital environment. The small black dots represent all sampled sites, while larger dots represent those sites where <i>Enterococcus faecium</i> strains were isolated. Genetically linked isolates [core genome single nucleotide polymorphisms (cgSNP) alleles ≤ 25] are depicted by dots with the same color.](https://brieflands.com/journals/jjm/articles/159860/figures/jjm-159860-i001-F1-preview.webp "A sketch map of sampling soil sites within the hospital environment. The small black dots represent all sampled sites, while larger dots represent those sites where <i>Enterococcus faecium</i> strains were isolated. Genetically linked isolates [core genome single nucleotide polymorphisms (cgSNP) alleles ≤ 25] are depicted by dots with the same color.")
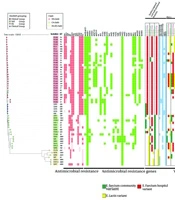
 distribution, and virulence factor gene variants. Isolate labels indicate their source: P for patients, S for soil. Clade assignments are represented by colored strips, while HierBAPS-based groupings are shown with different colored symbols. Dark red indicates resistance, and lighter red indicates intermediate resistance. The antibiotics tested include: Vancomycin (Van), teicoplanin (Teico), tigecycline (Tige), tetracycline (Tet), rifampicin (Rif), ciprofloxacin (Cipro), penicillin (Pen), linezolid (Line), chloramphenicol (Chlo), minocycline (Mino), levofloxacin (Levo), ampicillin (Amp), and erythromycin (Ery). Variants of virulence factor genes are boxed, with color codes: Green for the <i>Enterococcus faecium</i> community variant, red for the hospital variant, and yellow for the <i>E. lactis</i> variant.")
 based on core genome SNPs of 56 isolates. Each circle represents an isolate, with green circles representing soil isolates and red circles representing clinical isolates. The numbers between isolates show their core genome single nucleotide polymorphisms (cgSNP) distance. Isolates within the threshold of 25 alleles are considered genetically linked; gray zones were drawn to bridge these isolates.")

, soil (green), and <i>E. lactis</i> (brown).")