Some research has proved the effects of scorpion KTxs on some potassium channels consisting of SKCa, Kv1.x, BKCa, Kv11.1, and IKCa (
24,
25). However, new KTx inhibitors are required for channel structure-function studies as potential drugs for the treatment of potassium channel-related disorders for which an inhibitor does not exist (
25). Basic scorpion KTxs have been extensively employed in studies of toxin/potassium channel interaction assays. However, research on acidic KTxs has been progressed slowly (
32). The findings of the current study show how an acidic peptide, meuK2-2, derived from the scorpion M. eupeus venom, can be an effector of the potassium channel Kv1.3. To produce a trustworthy 3-D model of the meuK2-2/ Kv1.3 complex, a computational protocol employing homology modeling, followed by molecular docking experiments, pose clustering, and molecular dynamic simulations (100 ns), was developed. The results showed that meuK2-2 has a modified CSαβ structure, and when it encounters the Kv1.3 channel, it interacts with the residues of the turret and pore loop of Kv1.3; eventually, His9 penetrates the pore of the channel and blocks the ion permeation pathway. Moreover, the position of meuK2-2 as a lid above the Kv1.3 helps block the channel.
The vast majority of scorpion venom peptides are basic. However, a few anionic peptides with no toxicity properties have previously been characterized from the venom gland of
Mesobuthus martensii [BmKa1, BmKa2 (
33), and HAP-1 (
34)],
Tityus costatus (four peptides) (
35),
Scorpiops jendeki (SJE098.1 and SJE098.2) (
35),
Buthus occitanus Israelis (BoiTx776 from) (
36),
Hottentotta judaicus (HjVP) (
37), and
Androctonus bicolor (
38). The function of most of these peptides has not yet been characterized. It was previously thought that these peptides only play a part in balancing the pH of scorpion venom liquid (
33). However, Shi et al. recently showed that HAP-1 has an inhibitory effect on the antimicrobial peptide activity of M. martensii Karsch venom (
34). Now, acidic peptides are known as a new class of scorpion peptides characterized by (1) IP < 5.0; (2) being highly hydrophilic; and (3) having random coil regions and some α-helical domains (
34). Due to IP (4.42), the presence of random coil, and α-helical domains in the 3-D structure, meuK2-2 is an acidic toxin that can make it unique as a toxic KTx. This peptide appears to be a bifunctional peptide with two roles in the venom gland of M. eupeus, blocking the K channel and balancing the pH of the venom liquid.
The homology modeling of meuK2-2 revealed a CSαβ folding. Peptides with the CSαβ structure are the most predominant components of scorpion venom-affecting potassium channels (
39). A study using the molecular evolutionary analysis showed that most CSα/β toxin scaffolds underwent the periodic effect of positive selection, compared to non-CSα/β linear toxins (
40). KTxs are structurally classified into seven different groups, i.e., α, β, γ, κ, δ, λ, and ε-KTx. Seven different scaffold motifs have been identified within the structure of scorpion toxins: CS-α/β, CS-α/α, ICK (inhibitor cystine knot), DDH (disulfide-directed β-hairpin), Kunitz-type structural fold with a double-stranded antiparallel β-sheet flanked by an α-helix, and a knotting type fold, stabilized by four disulfide bridges (
41). The CSα/β motif occurs in α-, β-, and γ-KTxs subfamilies (
39). MeuK2-2 was also classified as an α-KTx, confirmed by the presence of the CSα/β motif in its structure.
In one classification, scorpion toxins were classified into six main groups and 16 subgroups, based on both functional and structural characteristics (
40). The CSα/β toxins were classified as the leading group with six subgroups, including CSαβ‘alpha, CSαβ’beta, CSαβ’chlorotoxin, CSαβ’lipo, CSαβ’scorpine, CSαβ’long-chain, and CSαβ'short-chain. Only the last three subgroups affect the potassium channels (
40). Excluding CSαβ’scorpine’, which contains β-KTxs, meuK2-2 is classified in the CSαβ’short-chain’ group because of its short length (28 amino acids).
Mutagenesis and computational experiments have suggested various interactions between potassium channels and scorpion toxins (
24). Our docking experiments determined that meuK2-2, like charybdotoxin (
42), is a pore blocker toxin that binds to the pore loop through a residue (Ser11), and like ErgTx1 (
43) and Cs1 (
19), two turret blocker toxins, binds to the turret through a hydrophobic residue (Ala28). Unlike Charybdotoxin that blocks the channel by entering the side chain of Lys residue in the pore, meuK2-2 occludes the pore with the His residue. Both Lys and His are classified in amino acids with positively charged side chains.
The importance of hydrophobic side chains in the stable binding of potassium channel blockers has previously been identified. Leneaus et al. demonstrated that hydrophobic side chains stabilize the binding of quaternary ammonium (QA) compounds (potassium channel blockers) to the surface of the potassium channel cavity (
44). Therefore, the hydrophobic interaction between meuK2-2 and Kv1.3 through Ala28 and Ser172 could play an essential role in stabilizing the meuK2-2/ Kv1.3 complex.
Some scorpion toxins like charybdotoxin did not induce conformational changes in binding to potassium channels (
42), while noticeable conformational changes were induced by some other scorpion toxins like kaliotoxin (
45) and ADWX-1 (
46). Conformational changes predominantly occur in potassium channels' channel turret or filter region upon different scorpion toxin bindings (
47). Computational simulations revealed that during the binding of ADWX-1 toxin to the Kv1.3 channel, the channel turret from one Kv1.3 chain makes close contact with ADWX-1 toxin, while the rest of the chains bend outward and move away from the ADWX-1 toxin (
46). These findings are in line with what was found here for meuK2-2. When meuK2-2 associates with the Kv1.3 channel, it binds to the turret and bends it inward close to the pore entry (
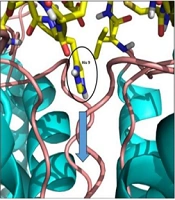
Figure 6).
In contrast to ADWX-1, the selectivity filter plays a vital role in binding the Kv1.3 channel to the scorpion toxin, maurotoxin. During the binding of maurotoxin to the Kv1.3 channel, conformational changes were induced in the channel filter region rather than the channel turret (
48). MeuK2-2, in addition to the turret channel, binds to the pore loop simultaneously. The binding of meuK2-2 to the pore loop induces conformational changes, leading to the selectivity filter getting tighter (
Figure 6).
In an overall comparison, meuK2-2 behaves somewhat like pore blocker toxins and somewhat like turret blocker toxins. Pore blocker toxins plug the K+ ion conduction pathway by entering a residue in the channel pore, whereas turret blockers locate as a lid at the pore entry and prevent K+ ions currents (
21). MeuK2-2 has both features in the interaction with Kv1.3. Moreover, a tighter selectivity filter results from the meuK2-2/ Kv1.3 interactions. We propose it as a new mechanism of scorpion toxins (or possibly basic scorpion toxins) and potassium channels interactions. Further research can shed new light on meuK2-2/Kv1.3 interactions.
The Kv channel blockers are known to be new tools to inhibit the activation of T-cells, resulting in specific immunosuppression. Besides, Kv1.3 is the primary channel on human effector memory T lymphocytes; consequently, it composes an appealing pharmacological target for autoimmune diseases mediated by T cell immunomodulation (
49). Functional characterization of meuK2-2 as a potent Kv1.3-targeted peptide will create new therapeutic agents for human autoimmune diseases.





 connect the β–sheet (yellow) to the alpha-helix (dark blue), facilitating the folding of the peptide into a CSαβ structure. Random coils are shown in light blue.")
 and Kv1.3 interactions. Residues of meuK2-2 and Kv1.3 contribute to the interaction; C, Kv1.3 channel blockade with the entry of residue His9. The pore path is indicated by a blue arrow.")
")
 and after MD-simulation (orange). The green arrow indicates the channel pore. Note the position of His9, before and after MD simulation.")

