1. Background
Breast cancer is the most common type of malignant tumor in women worldwide, excluding various biological characteristics and clinical behaviors, such as non-melanoma skin cancer (1). Based on current data from the american cancer society, 1,665,540 new cases and 585,720 deaths are expected to occur in the United States (US). In 2014, 235,030 new cases of breast cancer were reported in both sexes, and nearly 40,000 women died from the disease (2). Although many cytotoxic agents are used in the treatment of this cancer, their efficiency is somewhat limited. Thus, new treatments for breast cancer are in demand.
Rapamycin or sirolimus is a lipophilic antibiotic and fungicide produced by Streptomyces hygroscopicus, which was obtained from a soil sample on Easter Island in the late 20th century and developed clinically for its immunosuppressant characteristics (3). Rapamycin is now regarded as an anticancer drug that blocks mTOR by binding to FK 506-binding proteins (FKBP 12). The inhibition of mTOR reduces the phosphorylation and stimulation of S6K1 and also of 4E-binding protein 1 (4E-BP1), which helps to prevent the translation of mRNA required for cell cycle progression and cell proliferation (4). Reactive oxygen species (ROS) and cellular oxidative stress have long been correlated with cancer (2, 5). Oxidative stress might stimulate cancer cells, and receptor tyrosine kinase-activated cell cycle progression usually involves an increase in ROS signaling (6, 7). However, the antioxidant system in cancer cells is paradoxically activated to transform cells to produce a higher level of ROS in comparison with normal cells (8). In fact, several therapeutic agents promote cell death by increasing oxidative stress (9, 10). Anticancer drugs, such as doxorubicin, bleomycin, cisplatin, and paclitaxel, enhance oxidative stress (8, 11, 12). This common effect suggests that cancer cells are more susceptible to oxidative stress since they function with a raised level of ROS (13). Hence, the increase in the ROS level by these anticancer drugs pushes the cancer cells beyond the breaking point where cellular organelles and DNA are damaged and the cell undergoes apoptosis. Thus, a recurrence of the tumor after therapy likely results from a subset of cells that have developed the ability to overcome oxidative damage (14).
2. Objectives
Since few studies thus far have shown the effects of rapamycin on oxidative stress, the aim of the current study was to investigate malondialdehyde (MDA) and protein carbonyl levels as oxidative stress markers and also the GSH and ferric reducing ability of plasma (FRAP) to determine the antioxidant capacity induced by rapamycin in MCF-7 and MDA MB-231 human breast cancer cell lines.
3. Methods
3.1. Cell Lines and Cultures
MCF-7 and MDA-MB 231 cells were obtained from the Pasteur institute collection of cell cultures (Tehran, Iran). MCF-7 and MDA-MB231 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum, 2 mM glutamine, and 1% (v/v) penicillin and streptomycin at 37°C and 5% CO2 pressure. All of the cell culture reagents were obtained from Gibco-BRL (US).
3.2. Chemicals and Drugs
All chemical substances were purchased from Sigma (US). A stock of 1 mg/ml rapamycin was prepared by solving it in sterile dimethyl sulfoxide (DMSO) and maintaining it at -80°C. The ultimate vehicle concentration did not exceed 0.5% (v/v) either in the control or the treatment samples in all experiments.
3.3. Cell Treatment and Cell Proliferation Assays
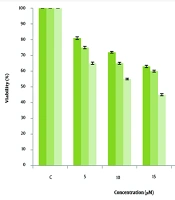
To test the effect of rapamycin on cell proliferation and select the appropriate rapamycin concentration, cells were seeded onto 96-well plates at 5 × 103 cells/200 μL/well. After an overnight incubation, triplicate wells were exposed with differing concentrations of rapamycin from 10 - 200 nM for 24, 48, and 72 hours. The relative percentage of metabolically active cells than the number of controls that received no treatment was then measured on the basis of the mitochondrial alteration of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT reagent) to formazine. The degree of MTT reagent that definitely changed to formazine indicated the amount of viable cells. The outcomes were analyzed in a 96-well format plate reader by measuring the absorbance at a wavelength of 570 nm. The percentage of metabolically active cells was compared with the percentage of control cells growing in the absence of rapamycin on a similar culture plate (2). The IC50 was determined by nonlinear regression analysis using the equation for a sigmoid plot. The MCF-7 and MDA-MB 231 cells were seeded in 50 cm2 flasks in duplicate. MCF-7 cells were treated (based on 50% inhibitory concentration (IC50) results) with 100 nM rapamycin for 72 hours. However, since the rapamycin was not effective on MDA-MB 231 cells, the same concentration was used for these cells. Untreated cells were considered the control group.
3.4. Preparation of the Cell Lysate
After incubation, trypsin was used to collect the cells; a 2-mL culture medium was added, and the mixture was centrifuged at 1500 × g for 5 minutes in a refrigerated centrifuge then washed with PBS (pH 7.4) three times. The pellet was transferred into an extraction solution, which contained 20 mM of a potassium phosphate buffer (pH 7) and a protease inhibitor cocktail. Cells were sonicated using a Biosonic IV sonicator for 3 min on ice and centrifuged at 20,000 × g for 50 minutes in a refrigerated centrifuge. The supernatant was used in the tests (2).
3.5. Measurement of Anti-oxidants and ROS Markers
3.5.1. Malondialdehyde (MDA) assay
MDA is the last marker of the lipid peroxidation pathway. This assay is according to the repercussion of MDA with thiobarbituric acid (TBA) that forms the MDATBA adduct that can be quantified calorimetrically and absorbs at 532 nm (3).
3.5.2. Carbonyl Protein Content Assay
Protein carbonyl is the marker of protein oxidation. The carbonyl was identified by measuring the protein carbonyl residues using dinitrophenylhydrazine (DNPH). Absorbance of the samples was measured at 370 nm (4).
3.5.3. Glutathione Reductase Assay
The reduction of 5, 5’-dithiobis (2nitrobenzoie acid) (DTNB) by GSH produced a yellow complex, which was used to indicate the level of thiol protein; the color intensity of 405 nm was proportional to the level of GSH (5).
3.5.4. Total Antioxidant Capacity Assay (TAC)
TAC was measured via the FRAP method. FRAP is a method that has been used to analyze the full potential of antioxidants in a sample. The absorbance of the samples was measured at 593 nm (6).
3.6. Statistical Analysis
All results were expressed as mean ± SD. Statistical analyses were carried out using PRISM 6 .0. Student’s t-test was utilized to analyze the statistical differences between groups under various conditions. A P value < 0 .05 was regarded as statistically significant.
4. Results
4.1. Cytostatic Activity of Rapamycin
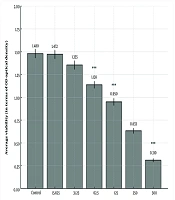
The rapamycin concentration that caused 50% growth inhibition was determined as the 50% inhibitory concentration (IC50). The IC50 value of rapamycin was 100 nM in MCF-7. The proliferation rate of MDA-MB 231 cells was not significantly different after treatment with rapamycin compared to the control cells (Figures 1 and 2).
 Was Significantly Decreased Compared to Control Cells (*P < 0.05)")
Figure 1.
The Cell Proliferation Rate of MCF-7 Cells After 72 Hours Incubation with Rapamycin at 100 nM (IC50 value) Was Significantly Decreased Compared to Control Cells (*P < 0.05)
")
Figure 2.
The Cell Proliferation Rate of MDA-MB231 Cells Treated with Rapamycin at 100 nM Showed no Significant Difference (P > 0.05)
4.2. Effects of Rapamycin on Antioxidants and Oxidative Stress Markers
The TBARS level in MCF-7 and MDA-MB 231 cells treated with rapamycin for 72 hours revealed a significant enhancement (P < 0.05) when compared to the control cells (Table 1). The same increase was also seen in the protein carbonyl levels in MCF-7 and MDA-MB 231 cells treated with rapamycin compared to the level found in control cells (Table 1). Moreover, a significant increase (P < 0.05) was observed in the GSH levels in the MCF-7 and MDA-MB 231 cells treated with rapamycin compared to the control group (Table 1). These results indicated that the level of FRAP in MCF-7 and MDA-MB 231 cells was significantly higher (P < 0.05) compared with the level in control cells (Table 1).
Table 1. Oxidative Stress Markers and Antioxidants in the Control and Rapamycin-Treated MCF-7 and MDA-MB 231 Cellsa
| Parameters | MCF-7 | MDA-MB 231 | ||
|---|---|---|---|---|
| Control | Rapamycin | Control | Rapamycin | |
| MDA (µmol/mg protein) | 0.02±0.004 | 0.08 ± 0.002b | 0.36±0.001 | 3.53±0.002b |
| Protein carbonyl content (µmol/mg protein) | 0.007±0.001 | 0.01±0.001b | 0.02 ± 0.002 | 0.03 ± 0.002b |
| GSH (µmol/mg protein) | 0.05±0.002 | 0.24±0.001b | 0.05 ± 0.001 | 0.22 ± 0.002b |
| Total antioxidant capacity (mmol/mg protein) | 0.05±0.001 | 0.17±0.002b | 0.02 ± 0.001 | 0.05±0.001b |
aThe results are expressed as mean ± SD for the four experiments.
bP < 0.05: significantly different compared to the controls.
5. Discussion
In this study, we evaluated the cytotoxicity effects of rapamycin on MCF-7 and MDA-MB231 breast cancer cell lines and assessed the level of oxidative stress. We found that rapamycin had the potential to induce oxidative stress, especially in the MCF-7 line. Enhanced stimulation of mTOR is identified in a variety of human cancers due to mutations in tumor suppressors and/or oncogene upstream regulators of mTORC1 functions. These mutations provide an advantage for cancer cells to selectively progress and proliferate in comparison with normal cells.
Rapamycin is considered one of the most promising drugs in cancer disease treatment and is currently being examined in different clinical trials (7-9). Preclinical studies have shown that T-cell leukemia, small cell lung cancer (SCLC), prostate cancer, and breast cancer have been the most susceptible cancers to rapamycin (10). The superoxide anion (O-2) is the principal free radical species produced through the usual aerobic metabolism and also may serve as a precursor for the production of other ROS. Moderate increases in O-2 levels actually perform an important task in mediating cellular proliferation (11). Numerous studies have confirmed that an increase in oxidative stress production via either ROS producers or antioxidant inhibitors may selectively execute tumor cells or possibly decrease tumor development and growth in several cancer cell lines. Therefore, ROS-manipulation strategies, which include approaches to remove or generate ROS in cancer cells, can potentially be effective treatments. Interestingly, cancer cells use ROS-scavenging systems as well as the transcription factor NF-E22-related factor 2 (NRF2) and its associated antioxidant systems to control their oxidative stress phenotype to avoid cell death (12).
ROS-mediated cancer cell proliferation was identified in liver, lung, breast, and many other types of cancers and can be avoided through the improvement of ROS-scavenging antioxidants (5, 13-16). In a number of cancers, the exogenous addition of H2O2 or endogenous oncogene-induced generation of ROS has been demonstrated to increase tumorigenicity and proliferation by activating the pro-tumorigenic and pro-proliferative signaling pathways, such as the MAPK/ERK and PI3K/AKT/mTOR pathways (17, 18). Ionizing radiation and chemotherapeutic components act together to promote ROS generation, thereby leading to irreversible oxidative injury.
Chemotherapeutic agents like anti-folates, vinca alkaloids, and taxanes enhance mitochondrial cell death via the release of cytochrome c and interrupt the mitochondrial electron transport chain, which results in enhanced superoxide generation. Other chemotherapeutics, such as doxorubicin, carboplatin, and cisplatin, noticeably increase ROS, which is the basis of their anticancer effects (12). A prior study investigated the sensitivity of all breast cancer cells to rapamycin and found that cell lines were cured with the administration of rapamycin at various concentrations for four days. Cell proliferation and DNA synthesis were then evaluated using MTT assay and thymidine incorporation, respectively. With the exception of MDA-MB-435 and MDA-MB-231, all cells were inhibited by rapamycin in all assays, and the decline in cell number was accompanied with a reduction in S phase progression (10). Additionally, a different study demonstrated the inhibition of the proliferation of the human pancreatic carcinoma cell line during treatment with rapamycin (19). When rapamycin and CC-5013 were combined, apoptosis was activated in MM cells (20). Our results revealed that rapamycin inhibited the proliferation rate in the MCF-7 cell line but was ineffective on the MDA-MB231 cell line. Other researchers have reported the same results using various drugs. The growth inhibitory features of temsirolimus (an analog of rapamycin) were evaluated in human breast cancer cell lines. The T-47D, MCF-7, and BT-474 cell lines are all estradiol responsive and were all strongly growth inhibited by temsirolimus. In addition, the growth of lines MDA-MB-468 and BT-549, which contained deletions of the PTEN tumor suppressor gene, was highly susceptible to being cured with temsirolimus. However, the MDA-MB-231 and MDA-MB-435 lines were resistant to treatment with temsirolimus (21). Another review obviously demonstrated that rapamycin exhibited inhibitory activity on the MCF-7 cell lines. Direct visualization via inverted microscopy revealed that the MCF-7 cell lines that were cured with rapamycin exhibited the properties of apoptosis as well as autophagy, vascularization, and cell shrinkage (22).
In vitro experiments revealed that rapamycin decreased 2’, 7’-dihydrodichlorofluorescein oxidation and enhanced glutathione. In this study, rapamycin inhibited tBHP-induced ROS generation. Cells cured with rapamycin had a higher viability in comparison to controls at 5 mM tBHP, and rapamycin efficiently protected human corneal endothelial cells (HCEC) from ROS-induced cell death by increasing levels of intracellular glutathione. These results indicated that rapamycin protects HCEC at high concentrations from oxidative injury-mediated cell death by the inhibition of ROS generation (23).
In other research, the molecular mechanism underlying partial rapamycin resistance in yeast was investigated. They used the yeast deletion collection to identify 15 deletion strains, resulting in a partial resistance to rapamycin. Among these were copper chaperone Lys7, superoxide dismutase 1 (SOD1), and copper transporter Ctr1, recommending a task for oxidative stress in rapamycin resistance. Increased levels of ROS specifically modified mTOR such that it no longer could completely bind with rapamycin: FKBP12. Therefore, increased oxidative stress modifies mTOR and prevents it from binding to the rapamycin: FKBP12 complex, finally resulting in its resistance to rapamycin (24).
mTOR regulation by ROS has been shown in both human cells and yeast. Chemically activated ROS results in the stimulation of mTOR in mammalian cells, and it is unclear whether this regulation is physiologically relevant (25, 26). In our study, lipid peroxidation expressed as TBARS was significantly enhanced in MCF-7 and MDA-MB231 cells treated with rapamycin compared with the control group. Other researchers have reported the same results with different drugs. For instance, enhanced lipid peroxidation was seen in MCF-7 cell lines treated with adriamycin and topotecan (2, 27). Additionally, in our research, we found a significant increase in protein carbonyl (PCO) levels in rapamycin-treated MCF-7 and MDA-MB231 cells in comparison with the control cells. In the same study with topotecan, the protein carbonyl levels increased in the MCF-7 cell lines compared with the control group (2). In the present study, we also found a significant rise in the GSH and FRAP activity as anti-oxidant markers in MDA-MB231 and MCF-7 cell lines were cured with rapamycin. This effect may be regarded as a response to the enhanced oxidative stress observed in cells cured with rapamycin.
It has been shown that doxorubicin raises the production of O-2 in isolated cardiac myocytes. SOD1 activity protects rat cardiomyocytes from producing intracellular ROS (28). Several studies have indicated that enhanced enzymatic and non-enzymatic anti-oxidant levels are responsible for the increased resistance to a number of chemotherapeutic agents (29). In another study, gene expression microarray analysis revealed that several ROS response genes were upregulated following rapamycin treatment, as well as glutathione reductase, delta aminolevulinate dehydratase, and SOD1 (30). The role of ROS in the proliferation of cancer has already been determined (31), and it remains to be examined whether it might serve as a predictor of rapamycin sensitivity in cancer malignancy. A reexamination of previous studies that have utilized in vitro cancer cell lines indicated that rapamycin resistance might possibly occur from the failure of rapamycin to bind with mTOR. It has also been demonstrated that rapamycin is less efficient at blocking the phosphorylation activity of mTOR in rapamycin-resistant MDA-MB-231 cell lines than with rapamycin-sensitive MCF-7 (32, 33). Other mechanisms of resistance to rapamycin have previously been described (34) The useful effects of rapamycin have largely been shown in preclinical animal models. However, the clinical success of rapamycin has been associated with only a few benign and malignant cancers. Numerous factors may lead to this moderate outcome in the clinic (35).
These findings support our hypothesis that rapamycin enhances the oxidative stress in MCF-7 and MDA-MB231 cells. In contrast, raising antioxidant levels after treatment with rapamycin might be one reason for the ineffectiveness of rapamycin in MDA-MB231 and also the development of resistance in MCF-7.