



1. Introduction
Focal dermal hypoplasia, also known as goltz syndrome or goltz gorlin syndrome, is a rare multisystem disorder with infinite variability, which was first described in 1962 (1). It is an X-linked dominant disorder; hence, 90% of the affected patients are females (2). The disorder is lethal in males, and few identified males surviving goltz syndrome are postzygotic somatic mosaics. The fundamental underlying defect is a mutation in the PORCN gene on the X chromosome (Xp11.23). This gene provides instructions for Wnt signalling, which is critical for the normal embryonic development of the skin, skeletal system, nails, hair, and eyes (3).
The lack of Wnt signalling results in the defective development and functioning primarily in the skin, skeletal system, and eyes in the cases of goltz syndrome (4). As few as only about 200-300 cases of goltz syndrome have been reported in the literature. We present one case of goltz syndrome referred to our institute with a giant cell tumor of the right distal fibula.
2. Case Presentation
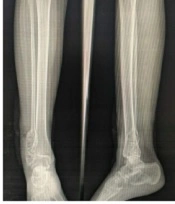
An 18-year-old female was referred to the Orthopaedics Outpatient Department with most complaints of pain and swelling over the right ankle lateral aspect for two months. On examination, there was a swelling of size approx. 11 × 7 cm on the lateral malleolus of the right ankle, which was tender, smooth, non-fluctuant, non-reducible in nature. The X-ray examinations revealed a typical soap bubble appearance (Figure 1). The patient was taken for resection of the mass, and a debridement sample was sent for histopathological examination when it was confirmed to be a giant cell tumor of the right distal fibula. The patient was admitted under department of orthopaedics and because of the presence of skin lesions, dermatology dept was consulted.

Typical soap bubble
The patient’s detailed history revealed that hyperpigmentation was present since birth. She also gave a history of a past surgery for ectrodactyly and syndactyly simultaneously for a split hand deformity and two or more webbed fingers, at the age of 5 years, respectively. The patient’s history also suggested chewing difficulties, poor weight gain, photosensitivity, gastroesophageal reflux, and vision problems.
A detailed examination revealed alternate light-dark patches in a linear blaschkoid pattern (Figure 2) on bilateral upper, lower limbs, and the trunk, pebbled skin texture, abnormally-shaped and peg-like teeth (Figure 3), malformed and ridged nails (Figure 4), thread-like dilated blood vessels on the cheeks (Figure 5), the sides of the nose show notching, facial asymmetry, the short stature of 140 cm, the weight of 42 kgs. Her right hand showed a scar mark on the dorsal aspect from the past surgery for ectrodactyly, and the absence of the right middle finger suggested oligodactyly (Figure 6). Ocular examination showed strabismus, left eye cataract, right anterior segment iris coloboma, right fundus disc coloboma with the tractional band, and right iris notching.

Trunk showing alternate light-dark patches in a blaschkoid pattern

Dental anomalies in the case of goltz

Malformed nails in the case of goltz

Telangiectasia on the patient’s cheeks

Oligodactyly and operated scar mark of ectrodactyly
A punch biopsy using a 5mm-punch was obtained from the left forearm lateral aspect of the hyperpigmented zone. The biopsy revealed hyperkeratosis and acanthosis in the epidermis. The dermis showed marked thinning with the periadnexal accumulation of adipocytes due to dermis thinning. These findings indicated focal dermal hypoplasia (Figure 7) (3).

Histopathological image of the case of goltz
The patient was diagnosed to be a case of focal dermal hypoplasia due to the following reasons: The presence of three major skin findings (namely congenital alternate light-dark patches over the extremities and trunk, congenital ridged malformed nails, telangiectasias) and three major skeletal manifestations (namely ectrodactyly, syndactyly, and oligodactyly) along with a large number of minor manifestations, including dental anomalies, hair shaft anomalies, pebbled skin texture, ocular defects, facial asymmetry, notching of sides of nares, photosensitivity, short stature, poor weight gain, gastroesophageal reflux, constipation, and giant cell tumor of bone (5).
3. Discussion
Goltz gorlin syndrome is an extremely rare multisystem disorder affecting females (6). A group of various manifestations, both major and minor, helped us to conclude the diagnosis of goltz syndrome. The prognosis is usually acceptable, with most individuals surviving in adulthood (7). Most of the anomalies are present at birth and remain unchanged after that. The diagnosis of cases with the most asymptomatic manifestations is delayed until later life, as it was in this case. The pain and swelling associated with the giant cell tumor brought the case to our notice; hence, the disorder could be clinically diagnosed and confirmed by dermatohistopathological examination.
According to the National Foundation for Ectodermal Dysplasia, three significant findings in Skin or Nails and one major finding in Limbs are a must in diagnosing goltz syndrome. The major findings are as follows:
Skin or Nails:
(1) Congenital patchy skin aplasia congenital patchy aplasia;
(2) Congenital alternate light-dark patches of skin color in a linear pattern;
(3) Congenital nodular fat herniation seen as soft, yellow-pink nodules on the trunk and extremities;
(4) Congenital small, ridged, or malformed nails; and
(5) Telangiectasias seen as thread-like lines on the face and trunk (8).
Limbs:
(1) Syndactyly- webbing of two or more fingers or toes;
(2) Ectrodactyly, known as a split hand-split foot malformation;
(3) Oligodactyly, ie, less than five digits on a hand or a foot;
(4) Transverse limb defect, ie, the congenital absence of hand, wrist, forearm, or elbow,
(5) Long bone reduction defect, ie, shortens the long bones in one or more extremities (9).
The minor findings are not included in the diagnostic criteria; however, theysupport the diagnosis of goltz syndrome.
Eye: Iris notching (50%), chorioretinal notching (60%), small or absent eyes, cataracts, involuntary eye movements, and tear duct obstruction (10);
Ectodermal manifestations in the cases: The patchy absence of the scalp hair or wiry hair (80%), wart-like papillomas (65%), dental anomalies such as missing teeth, peg teeth, and grooving or abnormal enamel (80%) (11), photosensitivity (40%), pebbled skin texture (58%), and hair shaft abnormalities (90%);
Skeletal: Scoliosis, small collar bones, giant cell tumors of the bones, and linear markings on the bones on X-rays;
Craniofacial: Facial asymmetry, small pointed chin;
Gastrointestinal symptoms: Short stature (65%), poor weight gain (77%), motor chewing and swallowing problems (41%), gastroesophageal reflux (24%), constipation (35%), and rarely abdominal wall defects;
Cognitive and psychological symptoms in the cases: Developmentally and intellectually normal (80 - 85%), emotional lability (50%), and withdrawn behavior (65%);
Neurological- rarely seizures and sensorineural hearing loss;
Cardiovascular: Truncus arteriosus;and
Obstructive sleep apnea in 93% of the cases.
