


1. Context
Cancer immunotherapy is a type of cancer treatment that uses the patient’s immune system to combat cancer. During the past two decades, various cancer immunotherapy products have been at the center of attention since they have been proven effective in the prevention, controlling, and elimination of various types of malignancies (1-3).
In the past decade, adoptive cell therapy (ACT) has changed the old face of cancer therapy. The ACT is described as transferring cells into a patient. The source of the cells used in ACT could be from the receiving patients themselves (autologous) or healthy third-party donors (allogeneic). Allogeneic ACT is considered advantageous in certain cases where the receiving patients do not meet the minimum criteria for using autologous cells due to the severity of the disease or the treatments they are receiving. Allogenic ACT overcomes the manufacturing barriers and difficulties of its autologous counterpart (Table 1). Furthermore, patients with particular oncological indications, such as chronic lymphocytic leukemia, suffer from T-cell dysfunction that might not be fully reversed during the manufacturing process of the adoptively transferred T-cells (4). This phenomenon has been known as a factor contributing to less favorable clinical outcomes following the T-cell therapy of the aforementioned subset of cancer patients (4).
| Variables | Advantages | Disadvantages |
|---|---|---|
| Autologous CAR T-cells | Low possibility of immune rejection occurrence; Eliminated risk of transferring pathogens from a third party donor | Variable end-product quality; Prolonged production time; High cost of production; Low number of starting population; Quality of the source cells can affect CAR T-cell therapy outcomes |
| Allogeneic CAR T-cells | Can be produced and made available as off-the-shelf products; Lower cost of production; Advanced manufacturing protocols can be utilized for their production; Guaranteed product quality, efficacy, and safety | Risk of host immune rejection; Risk of transferring pathogens from a third party donor; Shelf-life- and long-term storage-related limitations |
Some of the Advantages and Disadvantages of Autologous or Allogeneic Chimeric Antigen Receptor T-Cell Products
Genetically engineered T-cells with manipulated T-cell receptors (TCRs) and chimeric antigen receptor (CAR) T-cells are among famous examples of this type of therapy which have been investigated in numerous clinical trials with different types of malignancies (3). Genetically engineered T-cells harbor TCRs that recognize tumor-associated antigens (TAAs) or tumor-specific antigens (TSAs) on the surface of target tumor cells presented by the major histocompatibility complex (MHC). On the other hand, CAR T-cells, which are T-cells expressing CARs usually introduced to them via viral or non-viral methods, recognize their target TAA or TSA in an MHC-independent manner. With this background in mind, this review discusses the structure and functionality of CAR T-cells, their manufacturing process, their successful approval for the treatment of various hematologic malignancies, and the bright future ahead awaiting them.
2. CAR Structure and Activity
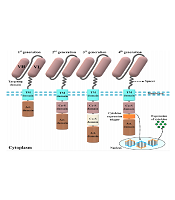
The CAR molecules consist of four different domains working together as a harmonized molecule providing the sufficient activation signal necessary for the activation of CAR T-cells upon target antigen recognition (Figure 1) (5). Conventional CARs comprise an extracellular domain (ECD), a hinge (alternatively known as a spacer), a transmembrane domain, and the necessary intracellular signaling domains (5). The ECD is responsible for the recognition of the target antigen of interest and triggering downstream CAR signaling cascades required for the activation of CAR T-cells. This primary signal passes through the abovementioned domains of CARs and eventually causes CAR T-cell activation (5, 6). Once activated, CAR T-cells enforce cytolytic reactions against the tumor cell of interest that results in its elimination (5, 6). Of note, to achieve (or even maintain) a successful anticancer treatment, CAR T-cells would have to eliminate tumor cells faster than they proliferate.
 molecule and four car generations developed by scientists throughout CAR T-cell therapy evolution. The targeting domain of the CARs represented in this figure is composed of a single-chain variable fragment derived from a conventional monoclonal antibody. First-generation CARs only harbored an activation domain; however, second- and third-generation CARs are designed to have one and two costimulatory domains, respectively. On the other hand, fourth-generation CARs are somehow second-generation CARs that have been designed to harbor a cytokine expression inducer. In detail, upon antigen engagement, the downstream signaling cascades result in the transcription and secretion of a cytokine of interest alongside the tumoricidal activity of the fourth-generation CAR T-cells. The aforementioned cytokine acts to improve the functionality of the CAR T-cells that secret it (VH, heavy chain variable domain; VL, light chain variable domain; Act. domain, activation domain; Co-S domain, costimulatory domain; TM, transmembrane).")
Different components of a chimeric antigen receptor (CAR) molecule and four car generations developed by scientists throughout CAR T-cell therapy evolution. The targeting domain of the CARs represented in this figure is composed of a single-chain variable fragment derived from a conventional monoclonal antibody. First-generation CARs only harbored an activation domain; however, second- and third-generation CARs are designed to have one and two costimulatory domains, respectively. On the other hand, fourth-generation CARs are somehow second-generation CARs that have been designed to harbor a cytokine expression inducer. In detail, upon antigen engagement, the downstream signaling cascades result in the transcription and secretion of a cytokine of interest alongside the tumoricidal activity of the fourth-generation CAR T-cells. The aforementioned cytokine acts to improve the functionality of the CAR T-cells that secret it (VH, heavy chain variable domain; VL, light chain variable domain; Act. domain, activation domain; Co-S domain, costimulatory domain; TM, transmembrane).
2.1. Extracellular Domain
The ECD of a CAR is composed of the target antigen-binding domain that is a crucial component for achieving the desired tumor-specific redirection of the engineered T-cells. The ECD is usually targeted toward a cell-surface antigen overexpressed in the cells of a specific type of malignancy. Therefore, its suitability can only be verified when the target TAA has a high expression rate on cancer cells and not on normal cells. The high expression rate of the CAR-targeted TAA on the surface of healthy cells can lead to severe adverse events known as "on-target off-tumor" toxicity (3, 7, 8). This toxicity can overshadow the selective tumor-targeting capability of CAR T-cells. To address this limitation, the TAAs toward which CAR T-cells are redirected should have a tumor cell-restricted expression alongside having a low level of expression on the surface of normal cells. The selection of a TAA with the aforementioned features is the first and foremost important step in designing CAR T-cells for fighting against a specific type of tumor.
The most frequent ectodomain used in the construct of CARs is derived from the single-chain variable fragment (scFv) of a TAA-specific monoclonal antibody (3). These scFvs can be of a human or murine source or can be the humanized version of a murine-derived scFv. The scFvs are made of a light chain and heavy chain linked by a flexible peptide linker that improves the affinity of the CAR molecule to target antigens. The scFvs obviate the need for tumor antigen processing and presentation by MHC molecules. This is where CAR T-cells act differently from endogenous T-cells or TCR therapies. In detail, TCR therapies demand target antigen processing and major HLA restriction for the activation of the engineered T-cells; however, CAR T-cells act independently of MHC.
The ECD is connected to the intracellular domains by an extracellular hinge domain and a transmembrane domain (composed of hydrophobic amino acids) (3). One of the downsides of using scFvs as the targeting domain of CAR T-cells is their spontaneous aggregation. This unwanted clustering results in the antigen-independent downstream singling of the scFv-equipped CAR T-cells that is known as “tonic signaling” (9). This occurrence leads to the exhaustion of CAR T-cells and consequently the abrogation of their antitumor capacity (9). One of the most potent and smartest strategies for overcoming this caveat is the incorporation of a VHH (camelid single-domain antibody fragments, also known as nanobodies®) into the CAR construct as the targeting domain (3, 10-16). The authors of the present study, as the first investigators in this field, have demonstrated that VHH-based CAR T-cells can be as potent as scFv-based CAR T-cells in terms of tumoricidal capacity (3, 10-16). Moreover, VHH-based CAR T-cells very well manage to overcome the limitations of scFv-induced tonic signaling (3, 10-16).
2.2. Hinge (Spacer)
The hinge or spacer is the linking domain present between the transmembrane domain and the ECD of CARs (17). It is commonly derived from the Fc portion of the immunoglobulin G subclass of antibodies (e.g., immunoglobulin G subclass 1 or immunoglobulin G subclass 4), immunoglobulin D, or CD8 domains. Several studies have demonstrated that the hinge is a fate-determining component in the CAR construct since it can affect the overall activity and the cytokine profiling of CAR T-cells (17-19). The CAR T-cell tumoricidal activity and persistence are also among other factors that may be enhanced using hinges with improved structures (19, 20).
2.3. Transmembrane Domain
The transmembrane domain of CARs acts as an anchor that helps keep the whole CAR construct in the membrane of the transduced T-cells. It also acts as a signaling entrance leading the target antigen engagement signal from the ECD of CARs to its intracellular domains. The transmembrane domains of CARs are usually derived from molecules, such as CD3-ζ, CD4, CD8, or CD28 (5).
2.4. Intracellular Domains and CAR Generations
The intracellular domains of CARs are responsible for the activation of CAR T-cells. The CAR T-cells are categorized into four different generations mostly based on their intracellular domains (21). The intracellular domain of modern-day CARs has an activation domain and one or two co-stimulatory domains (21). The activation domain is derived from Fcγ (i.e., the γ-chain from FcεRI) or CD3ζ (i.e., the ζ-chain of the TCR complex) (21, 22). The first-generation CARs only had an activation domain as their intracellular domain. These CAR T-cells demonstrated unsatisfactory in vivo expansion and tumoricidal activity and were unable to induce durable remissions (23). Later, researchers modified the construct of CARs by adding one or two costimulatory domains to their intracellular domain to generate second-generation and third-generation CAR T-cells, respectively (3, 5, 23). CD28, 4-1BB (or CD137), ICOS, and OX40 (or CD134) are among different molecules used as the costimulatory domain of CARs to date (3, 5, 23).
Studies have demonstrated that, in comparison to the first-generation CAR T-cells, the second- and third-generation CAR T-cells exhibit superior therapeutic efficacy due to their enhanced persistence, tumoricidal efficacy, and cytotoxicity and their mitigated differentiation and exhaustion state (3, 5, 23). The fourth-generation CARs, which are second-generation-based CARs, are called T-cells redirected for universal cytokine-mediated killing (24, 25). These CAR T-cells function as vehicles to produce and release a specific cytokine of interest inside the targeted tumor tissue mediating direct cytotoxicity alongside a second call for another immune recruitment (24, 25).
Apart from the abovementioned engineering methods used to improve CAR T-cell functionality, other tactics have also been used to achieve different aims (3, 5, 23, 26). For instance, bispecific CAR T-cells (e.g., CD19/CD22 CAR T-cells), tandem CARs, and suicide-switch-equipped CARs are among these strategies that can fill the empty spaces identified in various aspects of CAR T-cell therapy (3, 5, 23, 26, 27). Regarding suicide switches, equipping CAR T-cells with them enables their elimination upon feeling the need in the time of severe toxicities, such as cytokine release syndrome (CRS), neurological toxicities, or off-tumor adverse events affecting multiple organs (28, 29). A variety of such smart strategies are currently under clinical evaluation; nevertheless, other strategies have just been assessed in laboratories (all of which are comprehensively discussed elsewhere) (26, 28, 29).
3. CAR T-Cell Manufacturing, Conditioning Regimens, and Product Administrating
3.1. Manufacturing Process
Manufacturing CAR T-cells from autologous or allogeneic T-cells consists of several important steps, as shown in Figure 2. This process starts with leukapheresis that is performed to collect CD3+ lymphocytes (30-32). After isolating the desired lymphocytes from a patient’s or third-party donor’s blood, the remainder of the blood is returned to the patient’s or donor’s circulation (30-32). In the next step, these collected lymphocytes will be the target of genetic manipulation to express CARs on their surface (33). Gamma retroviral vectors, lentivirus vectors, and transposon/transposase systems are the three most frequently used methods for the genetic manipulation of T cells in CAR T-cell development (33, 34).
 molecule. Next, the developed CAR T-cells are expanded ex vivo to reach the desired dosage for infusion into the patients. Afterward, the manufactured CAR product is cryopreserved and then packed for shipping into the desired medical center in which the related patients are awaiting treatment. In the medical center, the product is thawed by professional staff and then intravenously infused into the patients. In the cases of allogeneic CAR T-cell products (i.e., “off-the-shelf”), the source of the T-cells used for the manufacturing of the desired CAR T-cells is from healthy donors rather than the patients themselves.")
Standard procedure of manufacturing a conventional autologous chimeric antigen receptor T-cell product. Blood samples are collected from the respective patients, and then T cells are collected from them. In a sterile environment, the isolated T-cells are genetically manipulated for the expression of the desired chimeric antigen receptor (CAR) molecule. Next, the developed CAR T-cells are expanded ex vivo to reach the desired dosage for infusion into the patients. Afterward, the manufactured CAR product is cryopreserved and then packed for shipping into the desired medical center in which the related patients are awaiting treatment. In the medical center, the product is thawed by professional staff and then intravenously infused into the patients. In the cases of allogeneic CAR T-cell products (i.e., “off-the-shelf”), the source of the T-cells used for the manufacturing of the desired CAR T-cells is from healthy donors rather than the patients themselves.
Moreover, lentiviral vectors are considered safer choices than gamma retroviral vectors because they tend to have safer integration target sites (into which the CAR gene fragment is inserted) (33). This is one of the reasons that lentiviral vectors are commonly used in clinics aiming to generate efficient CAR T-cells (35). Furthermore, these engineered T-cells are activated and expanded ex vivo. Several methods are utilized for the ex vivo activation of CAR T-cells (3, 36). Artificial antigen-presenting cells, antibodies targeting CD3, and CD3-CD28 targeting antibody-coated magnetic beads are among the different methods used for achieving this aim (3, 36). This step is also an important one in the whole process of CAR T-cell development since it has been shown to impact the antitumor efficacy of the resultant CAR T-cells. For example, it has been demonstrated that reducing the duration of the ex vivo expansion of CD19-redirected CAR T-cells augments their antileukemic efficacy (37). After reaching the desired therapeutic dose for the infusion, the manufactured CAR T-cells will be infused into the patient (mostly via the intravenous route).
3.2. Conditioning Chemotherapy
Most CAR T-cell therapy procedures are accompanied by conditioning regimens as they have proven to be contributing to better clinical results (3, 38-42). Fludarabine, cyclophosphamide, and bendamustine have been the most common chemotherapeutic agents used in relative clinical settings (3, 38-40). Conditioning regimens, as the name implies, contribute to the formation of a friendlier (or less hostile) environment for the administered CAR T-cells by the elimination of the patients’ lymphocytes and myeloid-derived suppressor cells (3). This phenomenon helps the adoptively transferred T-cells enforce more pronounced antitumor effects since they are exonerated from the sheer immunosuppressive effects of the host’s immune system (41, 43).
3.3. CAR T-Cell Infusion
The CAR T-cells are cryopreserved after generation and subsequently shipped to the relevant clinics/hospitals for infusion into the waiting patients. The US Food and Drug Administration (FDA)-approved CAR T-cells are administered via the intravenous route (a process taking about half an hour). However, preclinical findings (and clinical evidence) have demonstrated that the localized delivery of CAR T-cells into the desired tumor sites (also known as intratumoral administration) results in more pronounced antitumor responses and tumor rejection, compared to systemic administration, in animal models of solid human tumors (44-46). After the administration of CAR T-cells, the respective patients sometimes need to be carefully monitored since they might experience mild to severe toxicities that are dependent on various factors, including the patient’s disease burden, gender, or age and the adoptive cell dosage or the CAR T-cell product type (47, 48). In specific cases where toxicity grades are high, the related patients might be transferred to intensive care units where they might receive plasma exchange, mechanical ventilation, or other related therapeutics (e.g., tocilizumab, anakinra, lenzilumab, dasatinib, or metyrosine) in the cases of severe CRS, immune effector cell-associated neurotoxicity syndrome, hypoxia, pulmonary edema, and dyspnea (23, 47, 48). In case any CAR-T-cell-related toxicities arise, outpatients, such as those under the treatment of lisocabtagene maraleucel or tisagenlecleucel, are instructed to be accompanied by a close caregiver who can take them to a medical center for the appropriate toxicity management procedures in emergencies.
4. FDA-Approved CAR T-Cell Products
To date, four CAR T-cell products have entered the clinics for the treatment of patients with hematologic malignancies. All of these FDA-approved CAR T-cell therapies are CD19-redirected products making CD19 the most privileged and famous antigen of targeted cell therapy. Currently, more than 180 preclinical and clinical studies are investigating the antitumor efficacy and safety index of CD19-redirected CAR T-cells in different institutions. All this accentuates the therapeutic and financial importance of this antigen molecule, compared to that of other hematologic malignancy-associated target antigens, such as CD20, CD22, CD123, and BCMA (all of which come as the most investigated antigens after CD19). This section briefly highlights the details of each of the approved CAR T-cell products alongside investigating the clinical trials that led to their approval for medical use.
In 2017, Novartis’ tisagenlecleucel was the first CAR T-cell product to be granted approval by the US FDA for the treatment of relapsed/refractory (R/R) acute B-cell lymphoblastic leukemia (B-ALL) (49). Based on the 83% overall remission rate (achieved within 90 days following treatment) in a clinical trial with 63 R/R B-ALL patients, tisagenlecleucel was approved for medical use (49). Novartis has set the price of 475,000$ per tisagenlecleucel treatment, which is relatively high in comparison to that of other treatment modalities, such as chemotherapy or radiation therapy (49). Since both CRS and neurotoxicity might emerge in the relative patients following the administration of tisagenlecleucel, the product is sold with a black box warning (49).
In October 2017, Kite Pharma’s axicabtagene ciloleucel received FDA approval for the treatment of diffuse large B-cell lymphoma (DLBCL) patients who have relapsed or become unresponsive to two or more lines of prior treatments (50). Axicabtagene ciloleucel was granted permission for medical use based on a multicenter clinical trial in which a 51% complete remission rate was achieved in more than 100 enrolled participants (50). Similar to tisagenlecleucel, axicabtagene ciloleucel also comes with a black box warning regarding CRS and neurotoxicity since, in the second phase of the ZUMA-1 trial, 94% of the patients experienced CRS (more than 10% of whom required aggressive treatment) (50). Axicabtagene ciloleucel has been priced at 373,000$, making it slightly less expensive than tisagenlecleucel (50).
Furthermore, in July 2020, FDA approved Kite Pharma’s brexucabtagene autoleucel, under the trade name of Tecartus, for the treatment of adult patients with R/R mantle cell lymphoma under an accelerated approval pathway (51-53). This approval was based on the ZUMA-2 trial (NCT02601313) with 74 participants who had previously been treated with anthracycline- or bendamustine-based chemotherapy and a Bruton’s tyrosine kinase inhibitor but had not shown acceptable remission (51, 53). The complete remission rate after treatment with brexucabtagene autoleucel was reported to be 62% (51, 53). Furthermore, the objective response rate of this trial was also reported to be 87% (51, 53). Similar to the other FDA-approved CAR T-cell product of Kite Pharma, brexucabtagene autoleucel is also priced at 373,000$.
In February 2021, Bristol Myers Squibb’s lisocabtagene maraleucel was granted FDA approval for the treatment of R/R DLBCL patients who have undergone two or more lines of prior therapies (1). Based on a single-arm clinical trial with 73 and 54% of overall response rate and complete response rate, respectively, lisocabtagene maraleucel was granted permission for medical use (1). Similar to the other FDA-approved CAR T-cell products, patients undergoing treatment with lisocabtagene maraleucel have been reported to experience CRS, neurotoxicity, and protracted cytopenias (1). Bristol Myers Squibb has set a price of 410,000$ for lisocabtagene maraleucel per treatment that makes it a more expensive option in comparison to axicabtagene ciloleucel (1).
5. Concluding Remarks and Future Perspectives
The CAR T-cell therapy can serve as a great example of how basic and clinical sciences can work together to achieve extremely worthwhile aims. To date, this scientific phenomenon has shown promising results in many types of hematologic malignancies. This fact encourages researchers to try to also produce successful results regarding solid tumors. Furthermore, numerous studies are being conducted all over the world aiming to improve CAR T-cell function, persistence, stability, tumoricidal activity, and many other factors. Moreover, CAR-T-cell-therapy-associated toxicity, which still servers as an important boundary limiting the efficacy of this treatment modality, requires in-depth investigations to develop suitable mitigation strategies in this regard. There is almost no doubt that customizable and more intelligent CAR T-cells might have the crown for the future of cell-based immunotherapy.
