







1. Background
Helicobacter pylori is a Gram-negative spiral shaped bacterium that resides in the human stomach early in life and persists there for decades (1, 2). This bacterium is considered as the major cause of severe gastritis, peptic ulcers, mucosa associated lymphoid tissue (MALT) lymphoma, and gastric cancer (3, 4). The prevalence of infection is ranging from 25% in developed countries to more than 90% in developing areas. Pathogenesis of this bacterium depends on host and environmental factors as well as differences in expression of the bacterial virulence factors (5, 6). Genomic studies of the Helicobacter pylori strains from different countries have shown remarkable diversity in the gene content and arrangement of this bacterium around the world; however these genetic variations seem to be associated with geography and ethnicity. Genetic diversity of H. pylori strains within a population is depended on their allelic variations that may have driven through several events such as abounded mutations, genetic recombination, and a long evolutionary history of the strains (7). Accordingly, colonization of the human stomach with diverse strains, i.e. mixed infection, and also devolvement of quasispecies infection were established by several studies (8-10).
Among different methods for investigation of bacterial genetic diversity, MLST is an accurate sequence-based technique that can reveal genetic diversity and variability of H. pylori strains among different populations or within the strains isolated from each patient (11, 12). The population structure of H. pylori is clustered to hpEurope, hpAfrica1, hpAfrica2, hpEastAsia, hpNEAfrica (northeast Africa), hpSaul and hpAsia2. In Asia, H. pylori strains are also clustered into three subpopulations called hspEAsia (common in East Asia), hspAmerind (found in Native Americans), and hspMaori (isolated from Taiwanese, Aboriginals, Melanesians and Polynesians). The hpEurope cluster contained isolates from Europe, the Middle East, India and Iran (13).
Infection with H. pylori is a major concern in Iran with prevalence rates of 10% to > 80% in different provinces (14-20). Since Iran is surrounded by 12 countries and composed of more than 14 different ethnicities, it seems that genetically diverse populations of H. pylori strains are present in this country. However, there are not enough data to link these strains to each other and those from the other countries.
2. Objectives
In the current study, we aimed to determine the genetic diversity of H. pylori strains isolated from an Iranian population by using MLST method.
3. Methods
3.1. Study Population
A total of 37 H. pylori strains were isolated from biopsy specimens of patients whom referred to endoscopy unit at Taleghani Hospital during 2009 to 2010. Three gastric biopsies were obtained from each patient; one used for culture and the remaining two biopsies were used for rapid urease test and histological examination. A written informed consent was obtained from all patients under a protocol approved by the Ethical Review Committee of the Gastroenterology and Liver Diseases Research Center at Shahid Beheshti University of Medical Sciences.
3.2. Helicobacter pylori Culture and Identification
The specimens were immediately sent to the laboratory in a transport medium consisting of thioglycolate with 1.3 g/L agar (Merck, Germany) and 3% yeast extract (Oxoid Ltd, UK). Afterwards, the biopsies were carefully dissected and homogenized in brain heart infusion broth (BHI), and subsequently cultured on Brucella agar medium (Merck, Germany) supplemented with 10% FBS, 7% horse blood and a mixture of antibiotics, including vancomycin, trimethoprim, polymyxin B and amphotericin B (Sigma, Germany). The inoculated plates were incubated at 37ºC under microaerophilic conditions (5% O2, 10% CO2, and 85% N2) for 3 - 5 days. The grown colonies were characterized initially by colony morphology and Gram staining following positive reactions in biochemical tests, including oxidase, catalase and urease tests. Molecular identity of each strain was confirmed by PCR using genus and species specific primer pairs for 16srRNA and glmM as previously described (10). Pure cultures of the strains were stored at -70ºC in a preservative medium (BHI; fetal calf serum 15%, and glycerol 20%) for further studies.
3.3. DNA Extraction
To provide appropriate DNA extracts for MLST analysis, a single colony of H. pylori strains was randomly selected from each culture. Subcultures of the single colonies were prepared and lawn of the colonies were used for DNA extraction using QIAamp DNA extraction kit (QIAgen®, Hilden, Germany) according to the manufacturer’s instructions. Quality of DNA extracts was assessed by electrophoresis on 0.8% agarose gels and its concentration was measured using Nanodrop (NanoDrop™ 2000/c Spectrophotometers, Thermo Fisher Scientific, USA).
3.4. Phylogenetic Analysis of Helicobacter pylori Strains by MLST
We used MLST method to analyze genetic diversity of H. pylori strains. Seven housekeeping genes, included atpA, efp, mutY, ppa, trpC, ureI and yphC, were selected from H. pylori PubMLST database (http://pubmlst.org/helicobacter/info/primers.shtml) for sequencing under following conditions: All PCR amplifications were performed in 25 µL reaction mixtures containing 2.5 µL 10× PCR buffer, 0.3 µL of each primer (Table 1), 1 µL of template DNA (approximately 150 ng), 0.4 µL of dNTPs, 0.7 µL of MgCl2, and 0.2 µL Taq DNA polymerase. The reaction mixtures were cycled in an automated thermal cycler (Eppendorf, Hamburg, Germany) under the following conditions: initial denaturation at 94ºC for 4 min followed by 35 cycles of denaturation at 94ºC for 1 min, annealing at the indicated temperatures for each reaction in Table 1 (50ºC - 60ºC) for 45 s, extension at 72ºC for 1 min based on size of the expected fragment (1 min/kb), and a final extension at 72ºC for 10 min. The amplified genes were detected by electrophoresis in a 1.8% agarose gel. The primer sequences, annealing temperatures and the estimated size of the PCR products are summarized in Table 1.
Table 1.Oligoucleotide Sequences of the Seven Housekeeping Gene Loci Used for MLST Analysisa
| Target Gene | Primers (5’ → 3’) | Annealing Temperature, ºC | Amplicon, bp |
|---|---|---|---|
| atpA | F: GCTATTGGGCAAAAAGAATC | 56 | 790 |
| R: ACGACGCTGTATTCCATCGC | |||
| efp | F: GGCAATTTGGATGAGCGAGCTC | 56 | 548 |
| R: CTTCACCTTTTCAAGATACTC | |||
| mutY | F: GTGGTTGTAGYTGGAAACTTTACAC | 58 | 661 |
| R: CTTAAGCGTGTGTYTTTCTAGG | |||
| ppa | F: GGAGATTGCAATGAATTTAGA | 53 | 711 |
| R: GTGGGGTTAARATCGTTAAATTG | |||
| trpC | F: TAGAATGCAAAAAAGCATCGCCCTC | 58 | 642 |
| R: TAAGCCCGCACACTTTATTTTCGCC | |||
| ureI | F: AGGTTATTCGTAAGGTGCG | 52 | 707 |
| R: GTTTAAATCCCTTAGATTGCC | |||
| yphC | F: CACGCCTATTTTTTTGACTAAAAAC | 56 | 677 |
| R: TTTCTARGCTTTCTAAAATATC |
aPubMLST database (http://pubmlst.org/).
3.5. Data Analysis
Sequence datasets for seven housekeeping genes of the H. pylori strains from hpAfrica1, hpAfrica2, hpNEAfrica, hpEurope, hpAsia2, hpEastAsia, hspMaori, hspAmerind and hspEAsia populations were obtained from the PubMLST database (http://pubmlst.org/) as shown in Appendix 1 in Supplementary File. Sequence types (ST) of our 37 H. pylori strains were also determined after submission of nucleotide sequences of the MLST loci into H. pylori PubMLST database (http://pubmlst.org/helicobacter). For construction of the phylogenetic trees in order to compare the diversity of our strains with other populations around the world, all of the obtained sequence datasets were compared with our sequence data from 37 Iranian strains that were already assembled, trimmed and aligned by using CLC sequence viewer 7 (http://www.clcbio.com). The phylogenetic dendrograms were constructed using neighbor-joining clustering method. All the strains that showed a concatenated nucleotide sequence similarity ≥ 98% were selected for phylogenetic analysis.
4. Results
4.1. Nucleotide Diversity of the Housekeeping Genes and Allelic Similarity
A total of 37 H. pylori strains were obtained from 101 patients undergoing endoscopy in Taleghani Hospital, Tehran, Iran. Identity of all strains was confirmed by biochemical tests and PCR. Multiple sequence alignment analysis of the 37 Iranian H. pylori strains showed that nearly 38% (14/37) of the strains were highly similar in at least three gene loci. Among these strains two different sets, including 1470/1471 and 1439/1440/1441 were identical in all seven gene loci. Helicobacter pylori strain 1438 was seen to have five identical genes in common with 1439/1440/1441 set of the strains. Only one set containing two STs 1446 and 1465 had six identical allele types. In addition, two other different sets, including 1466/1469 and 1462/1467 were identical in four allele types. Helicobacter pylori strains 1452 and 1455 had only three alleles in common. Details of identical and similar H. pylori strains and corresponding STs are all highlighted in different colors in Appendix 1 in Supplementary File.
Our sequence analysis also revealed that ureI was the most identical allele (48.6%, 18/37), and after that the efp gene showed the highest similarity (40.5%, 15/37) in the studied population. The mutY/yphC alleles and atp/ppa/trpC alleles were found to be identical among 35.1% (13/37) and 32.4% (12/37) of the strains, respectively (Appendix 1 in Supplementary File). In addition, sequence analysis of MLST datasets from global populations showed that trpC was the most identical allele in all seven housekeeping gene loci examined. Moreover, sequence analysis also revealed that yphC was the most diverse among gene loci studied in 111 H. pylori strains from previously described populations and subpopulations worldwide.
4.2. Population Structure and Phylogenetic Analysis of Iranian Helicobacter pylori Strains
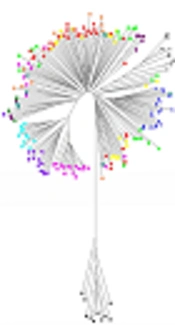
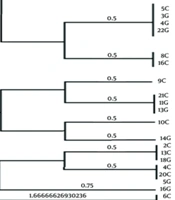
Neighbor-joining phylogenetic trees were constructed based on sequence datasets obtained from the seven housekeeping genes of 37 Iranian H. pylori strains and MLST datasets from 111 strains related to the Middle East and some neighboring countries, as well as global populations (Figures 1 and 2). These trees provided evidence for considerable diversity and specific geographic distribution and substructure within the Iranian H. pylori strains. The phylogenetic analyses also revealed that Iranian H. pylori strains clustered together with isolates from Turkey, Palestine, Israel, Lebanon, Egypt, Greece, Germany, Netherlands, Spain, UK, Finland, and Italy and are originally comparable to the ancestry of the hpEurope population.

Figure 1.
A, Neighbor-joining tree of 37 Iranian Helicobacter pylori isolates based on concatenated nucleotide sequences of 7 gene loci; B, neighbor-joining tree of 37 Iranian H. pylori isolates intermingled with 28 MLST datasets from neighboring countries. The Iranian MLST sequence IDs are shown in same numbers as shown in panel A.

Figure 2.
Neighbor-joining tree for detection of similarity of the Iranian Helicobacter pylori isolates with worldwide populations based on concatenated nucleotide sequences of 7 loci designed for MLST analysis. The Iranian MLST sequence IDs are shown in the numbers only.
5. Discussion
Helicobacter pylori is a fascinating human pathogen that chronically resides in the gastric epithelium of approximately half of the world population (21). These bacteria are extremely diverse and distributed in distinct geographic regions of the world (22). The genomic diversity within H. pylori strains is higher than most other bacterial species, and have long been used as a bacterial model for studying the microevolution of pathogens within a single host (23). In addition, it is believed that H. pylori strains have co-evolved within geographically defined populations, which can be used to explore the human migration routes. Helicobacter pylori is also well known for occurrence of wide single-nucleotide polymorphisms, homologous recombination, and genesis of pseudogenes through homopolynucleotide mutations within its genome (24). However, clonal clusters of H. pylori strains have also been identified from various ethnicities in different countries around the world (25-29).
Molecular fingerprinting of bacterial pathogens by exploiting MLST technique is currently one of the most popular genotyping methods, and has been used widely for characterizing several bacterial species. This method is based on DNA sequencing of typically 450 - 500 bp fragments to unravel allelic variations in multiple, usually seven, housekeeping genes (22, 23, 30, 31). Several studies have used MLST method for investigation of genomic diversity within H. pylori populations by using seven housekeeping genes, including atpA, efp, mutY, ppa, trpC, ureI, and yphC (22, 30, 31). Sequences of these gene markers are constrained due to importance of the functional proteins they encode. Therefore, they have proved to be suitable for analyzing genetic diversity of relevant H. pylori strains.
In this study, we investigated genetic diversity of 37 H. pylori strains isolated from Iranian patients suffering from different gastroduodenal disorders by using MLST method. We also included a dataset consisting of 111 H. pylori strains from 9 previously reported phylogeographic populations and subpopulations taken from PubMLST database. Results of sequence alignment analysis using MLST method revealed a high level of genetic diversity within most of the Iranian H. pylori strains. This finding is in agreement with a study conducted in Iran by Latifi-Navid et al., where they reproted that many pairs of Iranian populations are significantly different and moreover provided an indication that there was geographical structure to these variations (32). However, we also showed that about 38% (14/37) of the Iranian H. pylori strains analyzed in this study were similar in at least three gene loci. Interestingly, and based on the sequence analysis that was performed on the seven housekeeping genes used for MLST analysis we found the ureI gene as the most identical allele among Iranian H. pylori strains (48.6%, 18/37). Moreover, in a study performed on Malay population by Tay et al. (26), trpC was reported as the most diverse gene in all except the Malaysian Chinese population with the highest diversity about 7.6%, while the least diverse gene was atpA at 2.6% (28). In contrast, based on the multiple sequence alignments that we carried out on MLST datasets from different populations around the world, the trpC was found to have the least diversity among the examined genes.
In order to evaluate genetic diversity and clustering, we constructed three different phylogenetic trees by using sequence datasets of 37 Iranian H. pylori strains. Iranian strains were mixed with some strains from neighboring countries, and intermingled with 111 H. pylori strains from previously described populations and subpopulations worldwide, respectively (Figures 1 and 2). Our phylogenetic analyses showed that the Iranian H. pylori strains fall into distinct clusters, and were intermingled mostly amongst the hpEurope cluster, including isolates from Turkey, Palestine, Israel, Lebanon, Egypt, Greece, Germany, Netherlands, Spain, UK, Finland, and Italy (Figure 2).
In conclusion, our results showed similarity of nearly 38% of the Iranian H. pylori strains in at least three gene loci, while vast diversity was observed in other loci. Most of the strains were originally comparable to the ancestry of the hpEurope population. Further studies are needed to find a correlation between virulence potency of the strains and the characterized STs.