Several studies using rodent models have addressed the relationship between HFD and neurotrophic factors in the brain. Yu et al. Showed that BDNF and Trk β decreased following HFD in the hippocampus and ventromedial hypothalamic nucleus of male rats (
25). Furthermore, consuming a high fat refined sugar diet for 2 month decreased hippocampal BDNF and performance in the Morris water maze. They reported that the downstream effectors for the action of BDNF on synaptic plasticity, including synapsin I, CREB, and growth-associated protein 43 mRNA were reduced proportionally to BDNF levels (
7).
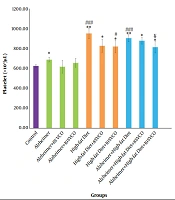
Inconsistent to the mentioned studies, the present study showed that administration of HFD for 9 months led to an increase in BDNF, Trk β (BDNF receptor), synapsin I (important for neurotransmitter release), and CREB (required for various forms of memory and is under regulatory control of BDNF). Furthermore, the results showed that the antioxidant could exaggerate HFD-induced increase in BDNF, Trk β, CREB, and synapsin I. It has been reported that vitamin C and E could exert protective effects by enhancing BDNF expression in stressed rats (
26). A study by Coskun et al. showed that vitamin C supplementation failed to protect the brain tissue against exercise-induced oxidative damage and behavior as a pro-oxidant (
27). In another study, vitamin E ameliorated the ethanol-induced changes on secretion of BDNF and neurotrophin-3 (
28).
In this study, HFD was used for 9 months, and longer than other studies. Such changes may be long-lasting changes, in which neurotrophic factors increase compensatory. Consistent with the current study, BDNF expression in the ventral tegmental area was increased by cocaine and they suggested that this may be essential for modulating cocaine-rewarding effects (
29).
In another study, rats, fed a diet rich in sugar, exhibited increased hippocampal inflammation (TNF-α and IL-1β mRNA) and oxidative stress, as indicated by an upregulation of NRF1 mRNA compared to control rats. In contrast, these markers were not significantly elevated in rats that received the cafeteria diet without added sucrose. Hippocampal BDNF mRNA was similar across all groups (
30), which confirms the current results.
The other finding of this study was attenuation of apoptosis following antioxidant administration in the HFD group. It has been reported that consumption of a fat-rich diet blunts leptin and insulin orexigenic signaling by a mechanism dependent on in situ activation of inflammation that could induce apoptosis of neurons and a reduction of synaptic inputs in the arcuate nucleus and lateral hypothalamus (
31,
32). Wang et al. showed that apoptotic hepatocytes were significantly greater in livers of rats fed HFD, and these were associated with a higher level of cleaved caspase-3 (
33). Caspase- 3 is one of the key executioners of apoptosis that is synthesized as inactive performs, which upon receiving an apoptotic signal, is cleaved and forms the active enzyme (
34). The ability of vitamin C or vitamin E to reduce cell damage elicited by various apoptotic stimuli has also been well studied (
35,
36). Vitamin C is an effective scavenger of hydroxyl radicals, and vitamin E is a lipid radical chain breaker that scavenges oxygen radicals and alkyl radicals. Taken together, it is possible that vitamin E and C inhibit ROS generation and might be implicated in the protection of HFD-induced neurotoxicity.
In conclusion, the current findings demonstrate that administration of vitamin E and C significantly attenuated HFD-induced neurotoxicity in the rat hippocampus. Therefore, it is likely that they may be useful as a potential treatment for the adverse effects associated with HFD.






, Trk (157 bp), CreB (156 bp), GAPDH (114 bp), and BDNF (180 bp) genes")
 of BDNF mRNA Concentration in Hippocampi")
 of Trk β mRNA Concentration in Hippocampi")
 of Synapsin I mRNA Concentration in Hippocampi")
 of CEREB mRNA Concentration in Hippocampi")
 of Caspase 3 Protein Concentration in Hippocampi")