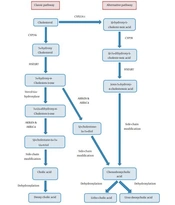
Familial intrahepatic cholestasis is a heterogeneous group of autosomal recessive liver disorders characterized by intrahepatic cholestasis, which can be divided into 3 main groups based on phenotypical differences: progressive familial intrahepatic cholestasis (PFIC), BRIC, and intrahepatic cholestasis of pregnancy (ICP). PFIC and BRIC can be further subdivided into 3 types by causative genes: (a) BRIC1/PFIC1, with mutations in ATP8B1 and a low/normal GGT level; (b) BRIC1/PFIC2, with mutations in ABCB11 and a low/normal GGT level; (c) BRIC3/PFIC3, with mutations in ABCB4 and a high GGT level (
4,
7).
The differentiation between BRIC and PFIC is based on phenotypic presentation: BRIC is characterized by intermittent recurrent cholestatic episodes, without evident liver damage, cirrhosis, and hepatic cell carcinoma. PFIC is progressive and could lead to end-stage liver diseases. It is thought that BRIC and PFIC are 2 ends of a continuum. In some cases BRIC could progress into PFIC, however, the inner mechanism is still unclear (
8,
9). All 3 causative genes of BRIC encode hepatocanalicular transporters: ATP8B1 encodes an amino-phosphlipid flippase translocating phospholipids from the outer to the inner leaflet of the plasma membrane, which helps protect the membrane from high bile salt concentration in canalicular lumen and to maintain its integrity; ABCB11 encodes the bile salt export pump (BSEP), which is the main exporter of bile acids from hepatocyte to canaliculi against a concentration gradient; ABCB4 encodes the multidrug resistance protein 3 (MDR3) functioning as a phospholipid floppase translocating phosphatidylcholine from the inner to the outer leaflet of the membrane, which is responsible for neutralizing the detergent effect of bile salt (
10,
11). Therefore, impair function of these genes would result in the injury of canalicular membrane of hepatocytes and biliary epithelium or bile canaliculi, leading to intrahepatic cholestasis.
Numerous mutations in ATP8B1 were detected in BRIC patients, however, not a single mutation was common among different populations. One missense mutation (c.1982T > C; p.Ile661Thr) is commonly detected in BRIC patients of western European origin. Roughly 50% of known ATP8B1 mutations identified to date are missense mutations. Putative splice site mutations, nonsense mutations, and small deletions have also been identified (
12). In the present case, 4 SNPs were detected in ATP8B1: c.696T > C, c.811A > C, c.2855G > A, and c.3454G > A (
Table 2). All of them were previously known as being related to intrahepatic cholestasis (
13,
14). Moreover, both c.2855G > A and c.3454G > A are nonsynonymous single nucleotide polymorphism (nsSNP), which are believed to cause phenotypic variety of human disease between individuals (
15). By considering that the conservation of amino acid residues in mammalian proteins homologous to ATP8B1, one can predict whether a missense mutation would lead to a mild disease phenotype or to a more progressive phenotype. Consistent with this opinion, point mutations in PFIC1 patients often affect conserved amino acid residues but the BRIC1 patients are on the contrary (
4).
ABCB11 is a member of the ATP binding cassette (ABC) superfamily (
11). There are also 4 genetic variations detected in our case. Both c.3084A > G, p.Ala1028 = (rs497692) and c.1331T > C, as well as p.Val444Ala (rs2287622) are known SNPs. The allele frequencies of rs497692 and rs2287622 in familial intrahepatic cholestasis were 67.2% and 74.5%, respectively. The c.3084A > G polymorphism could promote exon skipping and disrupt gene splicing, resulting in impair function of BESP. Similarly, the SNP variant A444 could reduce the levels of BESP protein compared with V444, which was also associated with drug-induced liver injury and intrahepatic cholestasis of pregnancy (ICP) (
16,
17). Importantly, 70A > T and c.1417G > A in our case were firstly reported in BRIC patient. The Mutation Taster prediction analysis indicated that both novel mutations are disease-causing-automatic and known to be deleterious. However, further study of these two mutations in the development of BRIC is still needed.
In our case, both ATP8B1 and ABCB11 genetic aberrations were detected. Which type of BRIC did the patient have? The common agreement is relied on which genetic variation is playing the predominant role during the disease process. Phenotypic characterization could help distinguish BRIC1 to BRIC2. ATPB1 is expressed in many tissues, abundantly in the pancreas, intestine as well as liver. The common extrahepatic features in ATP8B1 caused BRIC1, such as watery diarrhea, pancreatitis, and hearing impairment, which were absent in our patient. Thus, together with clinical presentations and genetic discoveries, we finally diagnosed the patient with BRIC2 (
18).
No medical intervention was consistently successful in BRIC to date. Cholestyramine has been used since BRIC was first recognized, although, without much success. In patients with ABCB4 deficiency, UDCA should be the first-line choice. However, the result of UDCA treatment in patients with ATP8B1 deficiency was controversial. Due to the fact that rifampin can generally relieve pruritus symptom in patients with cholestasis, it had also been considered in BRIC treatment. Use of rifampin could completely terminate an attack in some cases. When the patient’s liver enzyme was elevated at that time, rifampin was not in our treatment regimen for its liver toxicity. Surgical intervention like partial biliary diversion (PBD) and ileal exclusion have been successfully practiced in some BRIC patients. However, the permanent character of these operations makes them less suitable for patients with episodic BRIC. Temporary nasobiliary drainage (NBD) was effective in some BRIC patients. It could resolve pruritus and normalize bile salts in a short time. Liver transplantation was only considered in a few severe patients. Besides, plasmapheresis is also an option for the treatment of BRIC. Some studies reported the early use of plasmapheresis within 2 weeks after disease onset could shorten the duration of an attack, while other studies also had negative results (
2,
4,
19). In our case, the patient was conducted with PE within 2 weeks after his first onset, nonetheless, the TBIL still reached its maximum level after 8 times of PE treatments. In our observation, neither medication nor plasmapheresis interventions had effects on the natural process of BRIC, however, it could help alleviate the patients’ clinical symptoms.
It should be noticed that to date, evaluation of treatment efficacy in BRIC was only based on the single case report. Well-designed and multiple-centers prospective studies are needed, ideally with an intervention based on insight in the function of the proteins encoded by the causative genes. However, the limited patient number and the unpredictability of BRIC make it very difficult to design a randomized controlled trial (RCT). Therefore, single patient trial, also known as n-of-1 trial, could be a useful tool. N-of-1 trail is a multi-period crossover experiment comparing 2 or more treatments within 1 patient. It has been widely used in education and behavioral studies. Many physicians recognize that the nuances between different individuals and therapies should be tailored for the individual character, however, not only in a systematic manner across every patient. Therefore, n-of-1 trial, which focus on the objective determination of the optimal therapy for a single individual could help improve the clinical outcome of the patients. As the chronic, benign result, and recurrent episodic characters of BRIC, N-of-1 trail would be suitable for evaluating the treatments of this disease (
20).
In conclusion, we report a Chinese male with clinical approved and genetic diagnosis of BRIC. Both medication and plasmapheresis interventions could relieve the patient’s symptoms, however, neither could shorten the natural process of the disease. Two novel mutations in ABCB11 (c.70A > T, p.Lys24*, exon2 and c.1417G > A, p.Asp473Asn, exon13) were first detected and could help further understand the mechanism of BRIC development and design mutation-specific therapies for the patients in the future.










 and c&d (May 2016)")