Chemicals and reagents
All chemicals were of analytical grade and purchased from Sigma-Aldrich, Mo, USA.
Collection, identification, and extraction of plant material
Atriplex halimus L. aerial parts were collected from a village of Biskra state, Algeria and then identified by a botanist at the herbarium in the department of biology, University of El-Oued, Algeria. The samples were washed, dried at room temperature, and pulverized for extraction by the maceration method. Ten grams of the aerial parts of Ah powder was mixed with 100 mL distilled water at room temperature in the dark for 24 h. Then, the mixture was filtered through Whatman filter paper and then the filtrate was concentrated using a rotary evaporator and incubated at 40 °C to dry completely (
24). The percentage yield of the aqueous extract is 19.29 ± 0.24% of the dried sample (w/w).
Phytochemical Screening
Classical methods were used to identify the phytochemicals provides in the extracts of flavonoids (
25), saponins, cardiac glycosides (
26), terpenoids, mucilage (
27), alkaloids (
28), anthocyanins (
29), tannins (
30), coumarins, and carotenoids (
31).
Estimation of Total Phenolic
Total phenolic content was determined using the Folin-Ciocalteu method (
32). Two tenth milliliters of the aqueous extract of
Atriplex halimus was mixed with 1 mL of Folin-Ciocalteu reagent (diluted 1:10) and 0.8 mL of saturated sodium carbonate (75 g/L). After 2 h of reaction at room temperature in the dark, the absorbance at 765 nm was calculated. The tests were performed three times in order to guarantee the reproducibility of the results. The total phenolic content was expressed in mg equivalent of Gallic Acid (GAE) per g of sample.
Estimation of Total Flavonoids
The total flavonoid content of
A. halimus extract was estimated by the aluminum chloride colorimetric method (
33). One and half milliliters of a 2% AlCl
3 solution was added to 1.5 mL of sample or standard. After incubation for 30 min at room temperature, the absorbance was measured at 430 nm. Quercetin (Q) was used as a standard for plotting the calibration curve. The tests were carried out three times in order to ensure the reproducibility of the results. The results were represented in mg of Quercetin equivalent per g of sample.
In-vivo study
Animals and Handling
Thirty male Wistar albino rats, weighing 223.72 ± 5.37 g, were obtained from the animal house of the Pasteur Institute, Algeria. They were placed in animal’s house of the molecular and cellular biology department, the university of El-Oued, Algeria, in controlled (19 ± 1 °C) temperature, photoperiod (12 h light/12 h dark) cycle, and relative (64 ± 2%) humidity. Standard rat food and tap water were available ad libitum for the duration of the experiment. The rats were adapted for two weeks before tests under the same laboratory conditions. The experimental procedures were performed according to the National Institute of Health Guidelines for Animal Care and approved by the Ethics Committee of our Institution. The animals were randomly divided into five groups, each containing 6 rats as follow:
Groups 1: was serving as a control and received normal water;
Groups 2: was treated orally by gavage 200 mg/kg, b.w (3 days/week) aqueous extracts of Atriplex halimus (Ah) for 15 weeks;
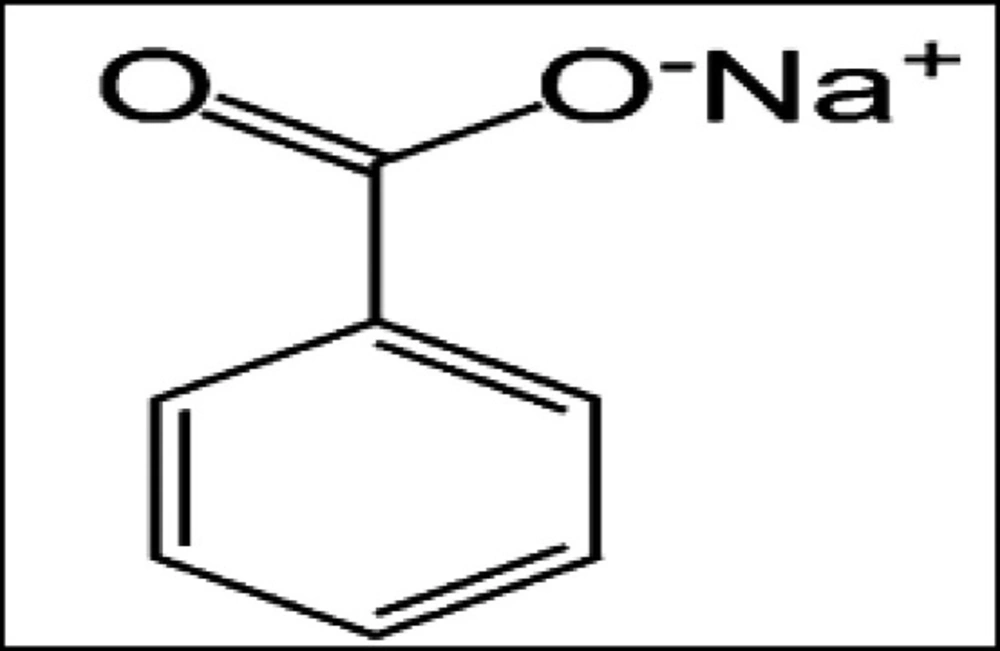
Groups 3: was received in drinking water 100 mg/kg, b.w/day of sodium benzoate (SB) daily for 15 weeks (
Figure 1);
Groups 4: was firstly treated with SB for 11 weeks and then treated curatively by 300 mg/kg, b.w/day of Ah (SB+AhC) for 30 days; Groups 5: was concomitantly administered Ah (200 mg/kg, 3 days/week) preventively with SB for 15 weeks (AhP+SB);
During treatment, body weight was recorded periodically during the experiment weeks.
Blood collection and preparation of tissue samples
At the end of the experiment, rats fasted for 16 h, sacrificed after anesthetized with chloroform by inhalation, and blood samples were collected. The serum was prepared by centrifugation and utilized for creatinine, urea, uric acid, albumin, and total protein concentrations and lactate dehydrogenase (LDH) activity assays. Blood glucose was measured by glucometer. The kidney was excised, rinsed, and weighed. Part of the tissue was stored at -20 °C for homogenate preparation. Another part of the kidney was transferred to fixative for histopathological examination.
Measurement of biochemical parameters
Serum creatinine, urea, uric acid, albumin, and total protein concentrations and LDH activity were measured using commercial kits obtained from Spinreact (Barcelona, Spain).
Antioxidants measurement
Preparation of homogenates (1)
Tissue kidney homogenates were prepared at 10% (w/v) in Tris buffer saline (Tris 50 mM, NaCl 150 mM, pH 7.4). The homogenate was centrifuged at 4000 revolution/min for 30 min at 4 °C. The supernatant was separated and used for the determination of oxidative stress markers.
Determination of Malondialdehyde (MDA) level (2)
MDA level was assayed according to the method described by Quintanilha
et al. (1982) (
34). 2 mL of MDA reagent (15% (w/v), trichloroacetic acid, 0.375% (w/v) thiobarbituric acid, and 0.25N hydrochloric acid) was added to aliquots of 1 mL diluted homogenate mixed with 20 µL of 2% (w/v) ethanolic solution of butylated hydroxytoluene. After incubating the mixture for 15 min in a boiling water bath and cooling in ice-cold, the precipitate was recuperated by centrifugation, and the absorbance was measured at 532 nm. MDA level was expressed as nmol of MDA/mg protein.
Determination of Reduced glutathione (GSH) level (4)
GSH was measured by the method of Ellman (1959) using DTNB (5,5′-dithiobis-(2-nitrobenzoic acid) reagent. 0.8 mL of kidney homogenate was mixed with 0.2 mL of 0.25% sulphosalicylic acid and the mixture was centrifuged at 1000 rpm for 10 min. Then, 0.5 mL of supernatant was added to 1 mL TBS (pH 7.4) and 0.025 mL (0.01 M) DTNB. The absorbance was recorded at 412 nm. The results were expressed as nmol GSH/g tissue (
35).
Determination of Glutathione-S-transferase (GST) activity (5)
GST activity was performed using the method of Habig
et al. (1974) (
36). To 2.8 mL phosphate buffer (pH 6.5), 25µL of kidney homogenates and 100 µL of (30 mM) GSH were added and the reaction was initiated by the addition of 100 µL (10 mM) CDNB (1-chloro, 2,4-dinitrobenzene). The absorbance was detected at 340 nm. GST activity was calculated in terms of μmol CDNB-GSH conjugate formed/min/g tissue.
Assay of Catalase (CAT) activity (6)
CAT activity was estimated according to Aebi (1984) method. The reaction was started by adding 200 μL of H
2O
2 (0.030 M) to 20 μL of supernatant and 780 μL of phosphate buffer (KH
2PO
4, 0.1 M, pH 7.5). The decomposition of H
2O
2 was monitored by following the decrease in absorbance at 240 nm every 30 sec for 2 min. The enzymatic activity was expressed in terms of international unit per minute and per gram of tissue (IU/min/g of tissue) (
37).
Protein level assay (7)
The protein content of kidney homogenates was determined as described by the Bradford method, and used bovine serum albumin as the standard (
38).
Histopathological study
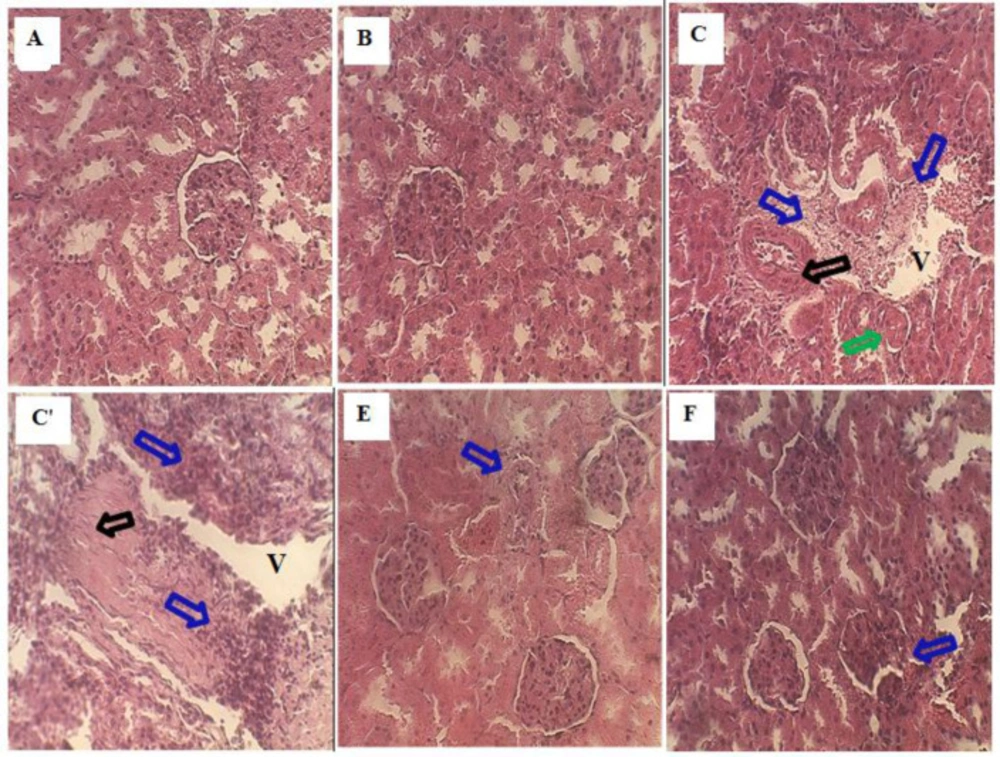
Part of kidney tissues of all experimental groups was immersed in fixative (10% formaldehyde solution) until the time of slice preparation. It was dehydrated in ascending graded concentrations of ethanol, cleaned with toluene, embedded in paraffin and sliced into 5 μm thick sections by a rotary microtome. Then, it was stained with hematoxylin and eosin. Histopathological evaluation was performed under a light microscope.
Statistical Analysis
Data were expressed as mean ± standard deviation (SEM) of six animals. Statistical analysis was carried out by using the Student t-test to compare means among the groups. Differences were considered significant at p < 0.05.