Characterization of PBDCNPE
Scanning electron microscopy was used to detect possible morphological changes on MWCNTs after the treatment. SEM images of MWCNTs and OCNT are shown in
Figure 1. The raw MWCNT being strongly entangled it is practically impossible to align them (
Figure 1a). As the oxidation proceeds the MWCNTs are gradually freed from the entanglements favoring their alignment. Furthermore the MWCNTs with small diameters are lost (
Figure 1b).
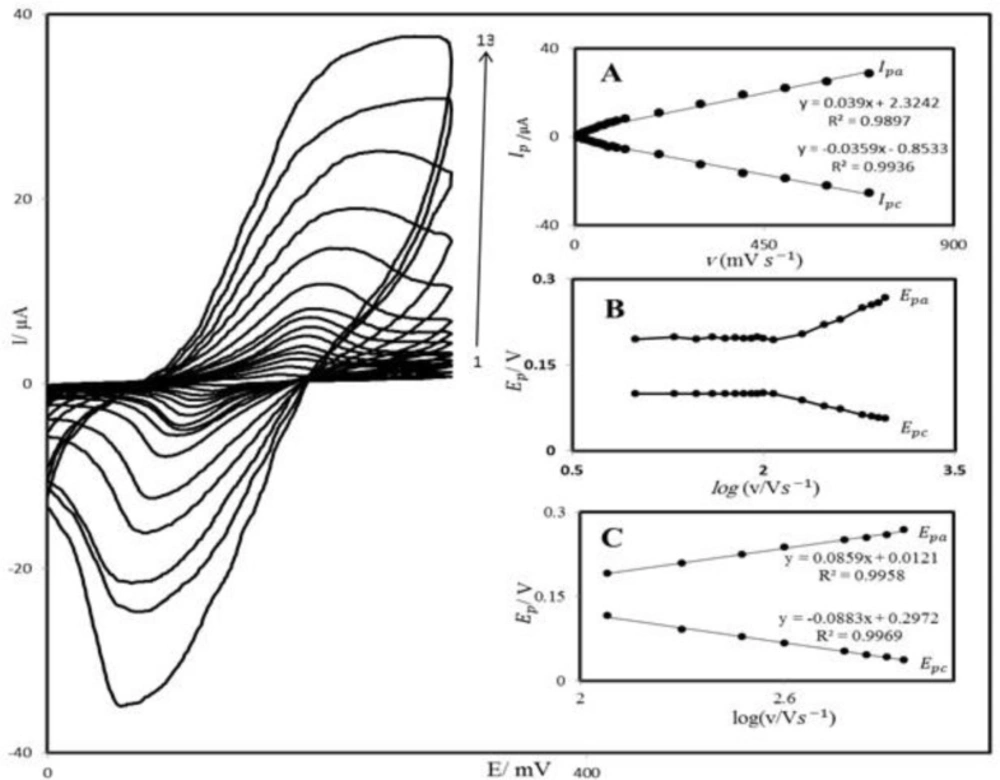
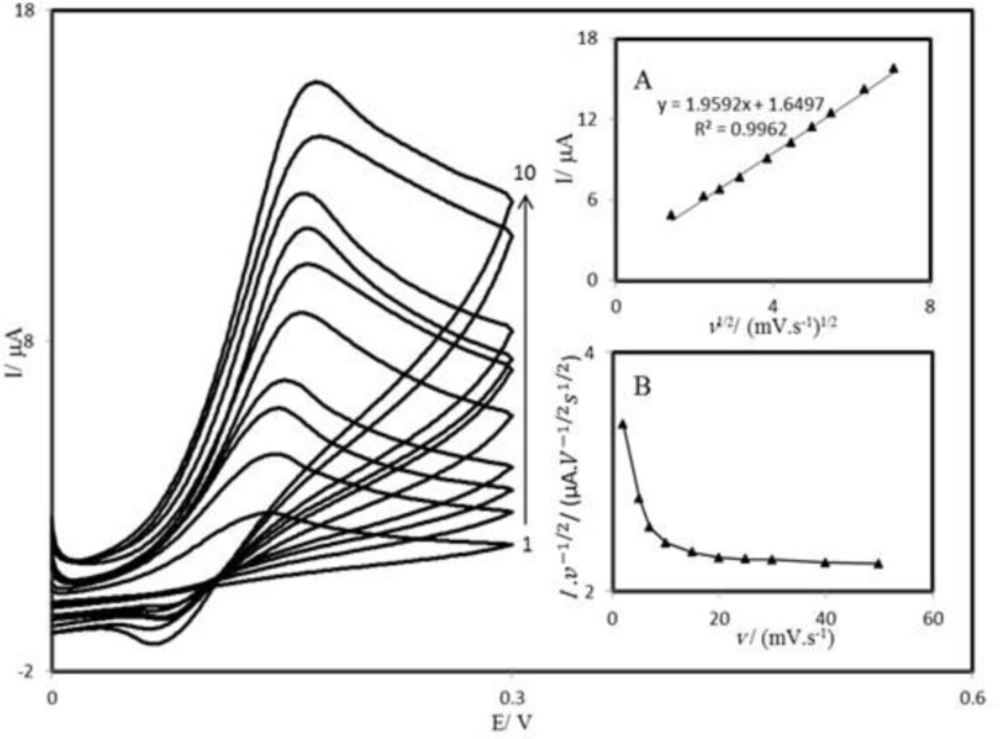
PBD compound is insoluble in aqueous media; therefore, we prepared PBDCNPE and studied its electrochemical properties in a buffered aqueous solution (pH 7.0) using cyclic voltammetry. The cyclic voltammograms for the modified electrode at different scan rates in 0.1 M phosphate buffer with pH 7.0 are shown in
Figure 2. A pair of reversible peaks are observed at E
pa = 0.20 V and E
pc = 0.10 V
vs. SCE and ΔE
p = (E
pa − E
pc) was 0.10 V. The electrode process was quasi-reversible, with ΔEp, greater than the expected for a reversible system. Inset A of
Figure 2 illustrates that the anodic and cathodic peak currents (I
p) were linearly dependent on υ at scan rates 10–1200 mV s
−1. A linear correlation was obtained between peak currents, and the scan rate indicates that the nature of redox process was controlled in a diffusion-independent manner (
Figure 2A).
An approximate estimate of the surface coverage (Γ) of the modified carbon paste electrode, given in mol cm
−2, was made by adopting the method used by Sharp
et al. (
38). According to this method, the peak current is related to the surface concentration of electroactive species, by the following Equation:
Ip = n2F2AΓv/4RT (1)
Where n represents the number of electrons involved in reaction, A (cm
2) is the surface area of the PBDCNPE, Γ (mol cm
−2) is the surface coverage and other symbols have their usual meanings. From the slope of anodic peak currents versus scan rate (
Figure 2B) the calculated surface concentration of PBD is Γ = 8.218 × 10
-8 mol cm
−2 for n = 2. Laviron (
39) derived general expressions for the linear potential sweep voltammetric response of surface-confined electroactive species:
log ks = α log (1 − α) + (1 − α) log α − log (RT/nαFυ) – α (1 − α) nαF∆Ep/2.3RT (2)
A plot of E
p as a function of log υ yields one straight line with a slope equal to 2.3RT/(1 – α)nF for the anodic peak (
Figure 2C). Using such a plot and Equation 2, the values of α and k
s were determined to be 0.31 and 1.17 s
-1, respectively.
Effect of pH on peak potential
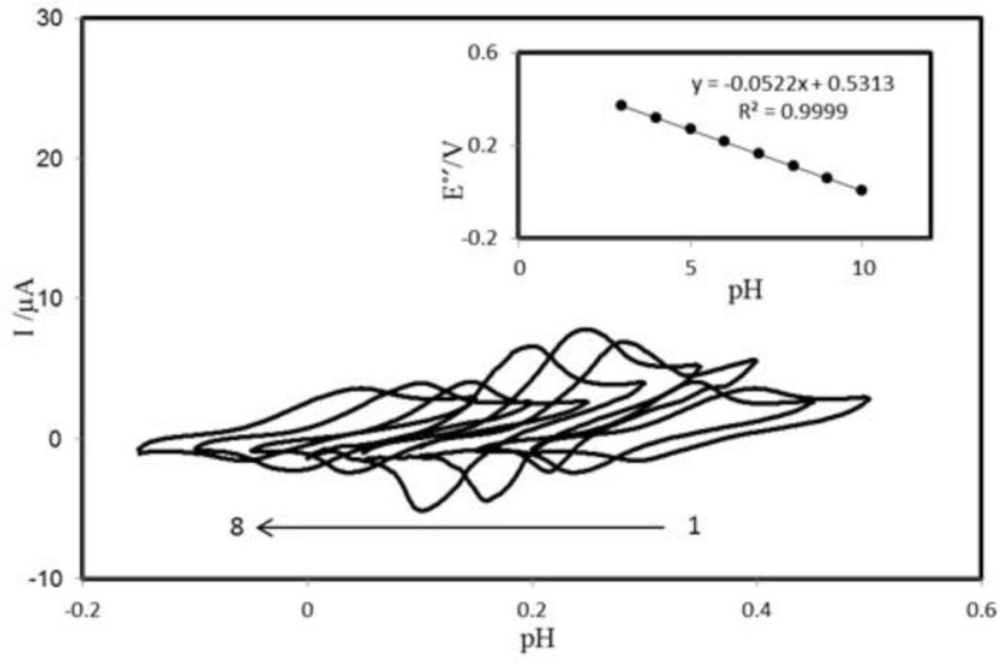
The voltammetric behavior of the PBDCNPE was characterized at various pHs by CV.
Figure 3 shows the CVs of the modified electrode in solutions at various pH values ranging from 2.0 to 10.0. The anodic peak potential was pH dependent. The inset of
Figure 3 shows Eº׳ as a function of pH. The results showed that the slope (Eº׳/pH) is -52.2 mV/pH units over a pH range from 2.0 to 10.0. This slope was close to the Nernstian value of -59 mV for a two-electron, two-proton process (
40). So two protons are transferred in the redox reaction in the pH range 2.0–10.0
Electrocatalytic oxidation of AA at a PBDCNPE
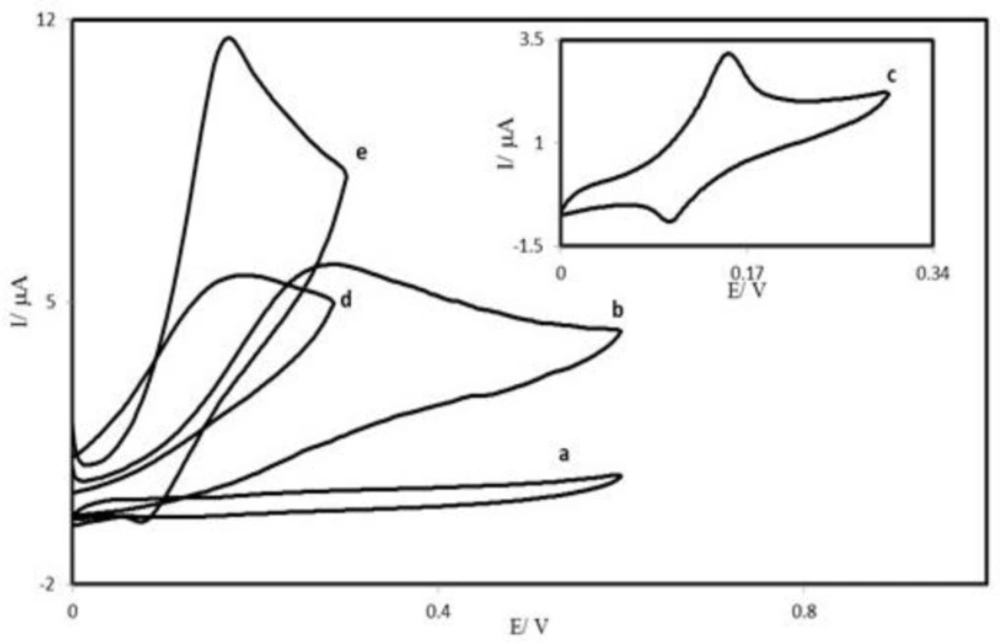
Figure 4 depicts the cyclic voltammetric responses from the electrochemical oxidation of 0.25 mM AA at the PBDCNPE (curve e), PBD modified CPE (PBDCPE) (curve d) and unmodified CPE (curve b). As can be seen, the anodic peak potential for AA oxidation at the PBDCNPE (curve e) and PBDCPE (curve d) was about 170 mV, while at the unmodified CPE, the peak potential was about 300 mV (curve b). From these results, it was concluded that the best electrocatalytic effect for AA oxidation was observed at the PBDCNPE (curve e). For example, the results show that the peak potential of AA oxidation at the PBDCNPE (curve e) shifted by about 130 mV toward negative values when compared with that at the unmodified carbon paste electrode (curve b).
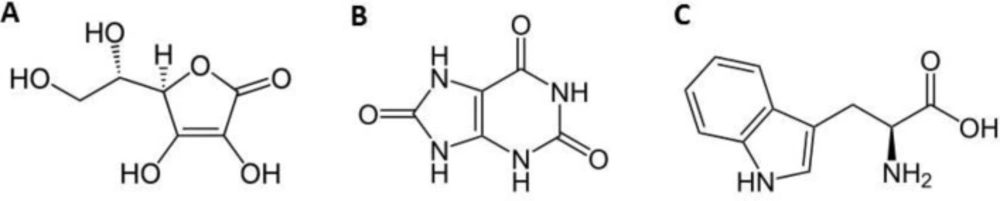
Structure of (A) Ascorbic Acid, (B) Uric Acid and (C) Tryptophan.
(a) SEM images of MWCNTs before oxidation and, (b) after oxidation.
Cyclic voltammograms of PBDCNPE in 0.1 M phosphate buffer (pH 7.0), at various scan rates: the numbers1–13 correspond to 10, 20, 50, 70, 80, 100, 120, 200, 300, 400, 600, 800 and 1200 mV s−1 scan rates, respectively. Insets: (A) Variations of Ip versus scan rates (B) Variation of Ep versus the logarithm of the scan rate. (C) Magnification of the same plot for high scan rates.
Cyclic voltammograms (at 100 mV s−1) of PBDCNPE at various buffered pHs. The numbers 1–8 correspond to 3, 4, 5, 6, 7, 8, 9 and 10 pHs, respectively. Inset: Plot of E°′ vs. pH
Cyclic voltammograms of: (a) an unmodified CPE in 0.1 M phosphate buffer (pH 7.0) solution and (b) the same electrode in 0.25 mM AA, pH 7.0 solution. (c) as (a) for PBDCNPE. Also, (d) and (e) as (b) at the surface of PBDCPE and PBDCNPE respectively
Cyclic voltammograms of a PBDCNPE in 0.1 M phosphate buffer (pH 7.00) containing 1.0 mM AA at different scan rates; the numbers 1 to 10 correspond to 2, 5, 7, 10, 15, 20, 25, 30, 40 and 50 mV s–1 scan rates, respectively. Insets: (A) Variation of the electrocatalytic currents vs. the square root of scan rate, (B) variation of the scan rate normalized current (Ip/v1/2) with scan rate.
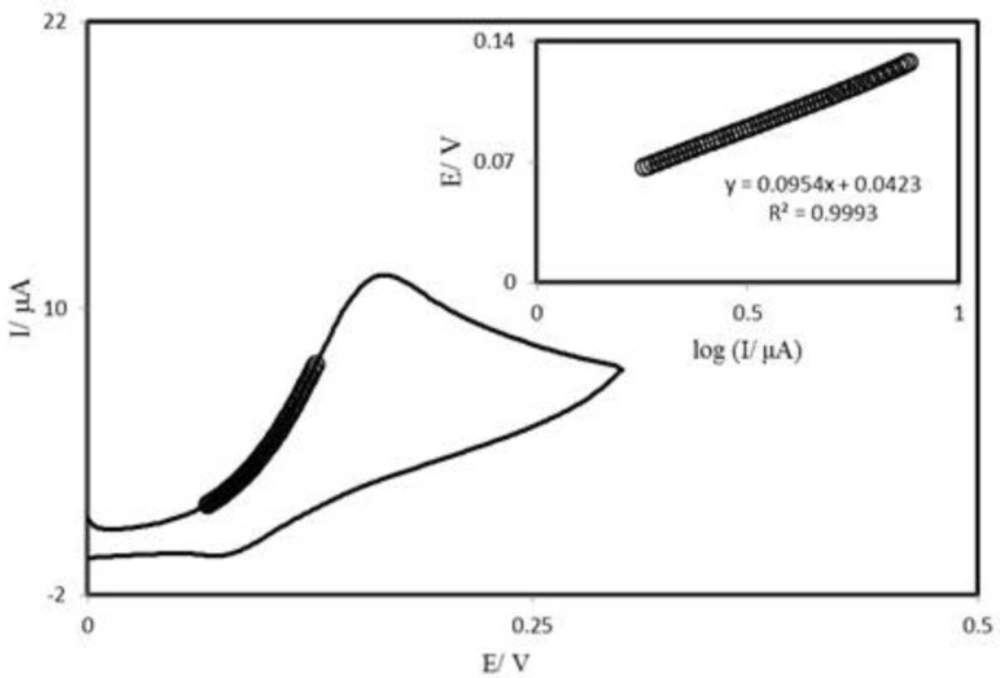
Cyclic voltammogram (at 25 mV s−1) of a PBDCNPE in 0.1 M phosphate buffer (pH 7.0) containing 0.25 mM AA. The points are the data used in the Tafel plot. The inset shows the Tafel plot derived from the cyclic voltammogram.
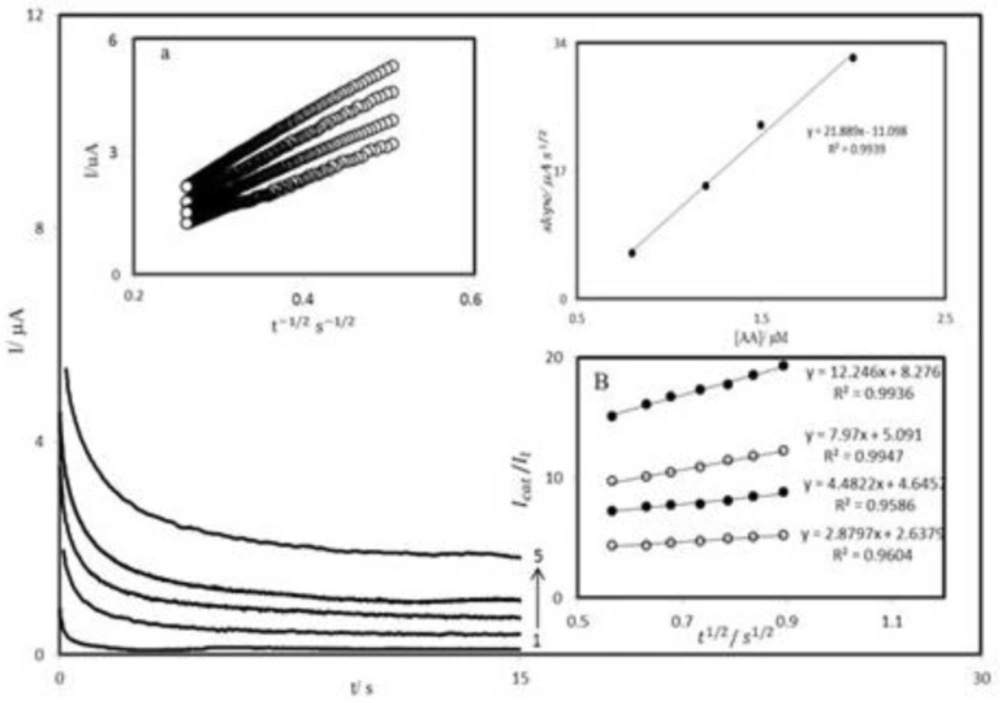
A chronoamperograms obtained at PBDCNPE in 0.1 M phosphate buffer solution (pH 7.0) for different concentration of AA. The numbers 1–5 correspond to 0.0, 0.8, 1.2, 1.4 and 2.0 mM of AA. Insets: A (a) plots of I vs. t-1/2 obtained from chronoamprograms 2–5 and A (b) plot of the slope of the straight lines against the AA concentration. B Dependence of Icat⁄Il on t1/2 derived from the data of chronoamprograms shown in a.
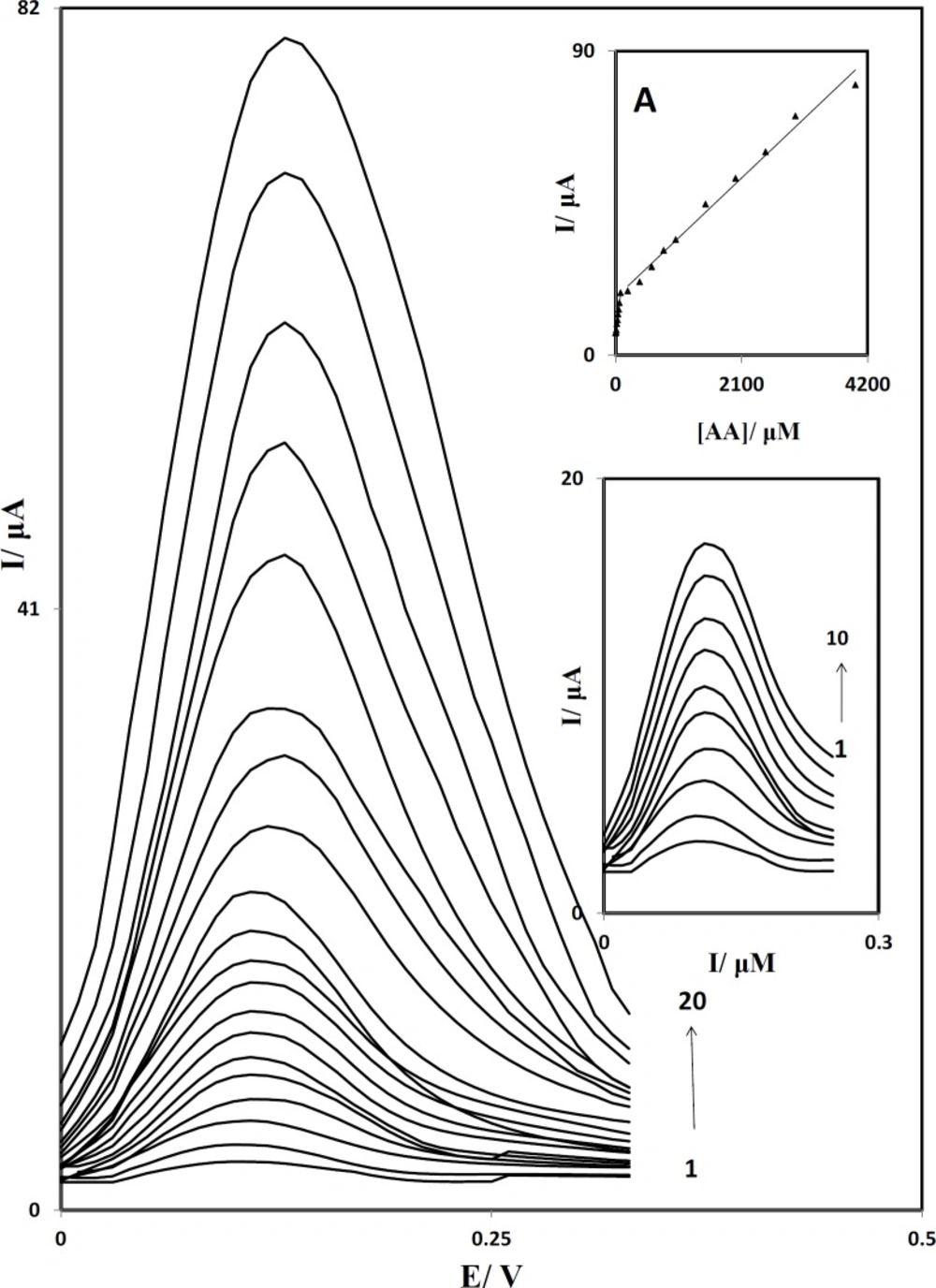
Differential pulse voltammograms of PBDCNPE in 0.1 M phosphate buffer solution (pH 7.0) containing different concentrations of AA. The numbers 1–20 correspond to: 1.0, 2.5, 5.0, 10.0, 20.0, 30.0, 40.0, 50.0, 60.0, 80.0, 200.0, 400.0, 800.0, 1000.0, 1500.0, 2000.0, 3000.0 and 4000.0 µM of AA. Inset A: The plots of the electrocatalytic peak current as a function of AA concentration. Inset B: Differential pulse voltammograms in the range of 1.0 to 80.0 µM.
Differential pulse voltammograms of PBDCNPE in 0.1 M phosphate buffer solution (pH 7.0) containing different concentrations of AA, UA, and Trp (from inner to outer) mixed solutions of 50.0 + 58.31 + 50.0, 100.0 + 116.63 + 100.0, 200.0 + 233.3 + 200.0, 400.0 + 466.5 + 400.0 and 600.0 + 700.0 + 600.0 respectively, in which the first value is the concentration of AA in μM, the second value is the concentration of UA in μM, and the last value is the concentration of Trp in μM. Insets: plots of the peak currents as a function of (A) AA, (B) UA, and (C) Trp concentration, respectively.
| Sample | Species | Added (µM) | Found (µM) | Recovery (%) |
|---|
| Vitamin C tablet | AA | 0 | 23.5 | - |
| | 10 | 33.1 | 96 |
| | 30 | 53.9 | 101.3 |
| Urine | UA | 0 | 9.5 | - |
| | 50 | 60.6 | 102.2 |
| | 70 | 78.8 | 99 |
| Serum | Trp | 0 | ND | - |
| | 50 | 51.4 | 102.8 |
| | 70 | 68.5 | 97.8 |
Similarly, when comparing the oxidation of AA at the PBDCPE (curve d) and PBDCNPE (curve e), a dramatic enhancement of the anodic peak current at the PBDCNPE relative to that obtained at the PBDCPE was observed. In other words, the data clearly show that the combination of carbon nanotube improve the characteristics of AA oxidation. The PBDCNPE, in 0.1 M phosphate buffer (pH 7.0) and without AA in solution, exhibited a well-behaved redox reaction (curve c), after addition of 0.25 mM AA, there was a dramatic enhancement of the anodic peak current (curve e), indicating a strong electrocatalytic effect (
41).
Effect of scan rate
The scan rate dependence of cyclic voltammograms for the PBDCNPE in 0.1 M phosphate buffer solution containing 1.0 mM AA is presented in
Figure 5. It can be noted from
Figure 5 that, with an increasing scan rate, the peak potential for the electrooxidation of AA shifts to more positive potentials, suggesting a kinetic limitation in the reaction between the redox sites of PBD and AA.
A plot of peak height (I
p) against the square root of scan rate (v
1/2), in the range of 2–50 mV s
-1, was constructed (
Figure 5A), which was found to be linear, suggesting that at sufficient overpotential the process is diffusion rather than surface controlled. A plot of the sweep rate normalized current (Ip/v
1/2) versus sweep rate (
Figure 5B) exhibits the characteristic shape typical of an EC
׳cat process.
Tafel plot was drawn from data of the rising part of the current–voltage curve recorded at a scan rate of 25 mVs
−1 (
Figure 6). This part of voltammogram, known as Tafel region, is affected by electron transfer kinetics between substrate (AA) and surface confined PBDCNPE, assuming the deprotonation of substrate as a sufficiently fast step. In this condition, the number of electron involved in the rate determining step can be estimated from the slope of Tafel plot. A slope 0.114 Vdecade
−1 is obtained indicating a one electron transfer to be rate limiting assuming a transfer coefficient of α = 0.52.
Chronoamperometric measurements
The chronoamperometry was employed along with other electrochemical methods for the investigation of electrode processes at chemically modified electrodes.
Figure 7A shows chronoamperometric measurements of AA at PBDCNPE. This figure represents the current–time profiles obtained by setting the working electrode potential at 350 mV for various concentrations of AA. In chronoamperometric studies, we have determined the diffusion coefficient of AA at PBDCNPE. For an electroactive material (AA in this case) with a diffusion coefficient of D, the current for the electrochemical reaction (at a mass transport limited rate) is described by the Cottrell Equation (
41):
I= nFAD1/2Cbπ-1/2t-1/2 (3)
Where D and C
b are the diffusion coefficient (cm
2 s
-1) and the bulk concentration (mol cm
-3), respectively. Under diffusion control, a plot of I versus t
-1/2 will be linear, and from the slope the value of D can be obtained.
Figure 7A inset a, shows the experimental plots with the best fits for different concentration of AA employed. The slopes of the resulting straight lines were plotted versus the AA concentration (
Figure 7A inset b). The mean value of the D was found to be 4.5×10
-8 cm
2 s
-1. Chronoamperometry can also be employed to evaluate the catalytic rate constant, k, for the reaction between AA and the PBDCNPE according to the method of Galus (
42):
I
C⁄I
L = γ
1/2[ π
1/2 erf(γ
1/2) + exp(-γ)/ γ
1/2] (
4)
where IC is the catalytic current of AA at the PBDCNPE, IL the limited current in the absence of AA and γ = kCbt (Cb is the bulk concentration of AA) is the argument of the error function. In the cases where γ exceeds 2, the error function is almost equal to 1, and therefore, the above equation can be reduced to:
I
C⁄I
L = π
1/2γ
1/2 = π
1/2(kC
bt)
1/2 (
5)
where t is the time elapsed (s). The above equation can be used to calculate the rate constant of the catalytic process k. Based on the slope of the I
C⁄I
L versus t
1/2 plot; k can be obtained for a given AA concentration. Such plots obtained from the chronoamperograms in
Figure 7A are shown in Figure 7B. The value of k explains as well as the sharp feature of the catalytic peak observed for catalytic oxidation of AA at the surface of PBDCNPE. Finally, the heterogeneous rate constant of catalytic reaction was calculated as k = 9.3 × 10
-1 cm s
-1.
Differential pulse voltammetry
Differential pulse voltammetry (DPV) has a much higher current sensitivity and better resolution than cyclic voltammetry, there for we used Differential pulse voltammetry to determine the concentration of AA. In addition, the charging current contribution to the background current, which is a limiting factor in the analytical determination, is negligible in DPV mode.
Figure 8 shows the differential pulse voltammograms obtained for the oxidation of different concentrations of AA at the PBDCNPE. The dependence of the peak current on the AA concentration is shown in inset A of
Figure 8 in the range of 1.0 to 4000.0 µM. This inset clearly shows that the plot of peak current versus AA concentration is constituted of two linear segments with different slopes, corresponding to two different ranges of substrate concentration. The decrease of sensitivity (slope) in the second linear range is likely to be due to kinetic limitations. Inset B shows differential pulse voltammograms in the range of 1.0 to 80.0 µM. From the analysis of this data, we estimate that the lower limit of detection of AA is of the orde of 0.3 μM.
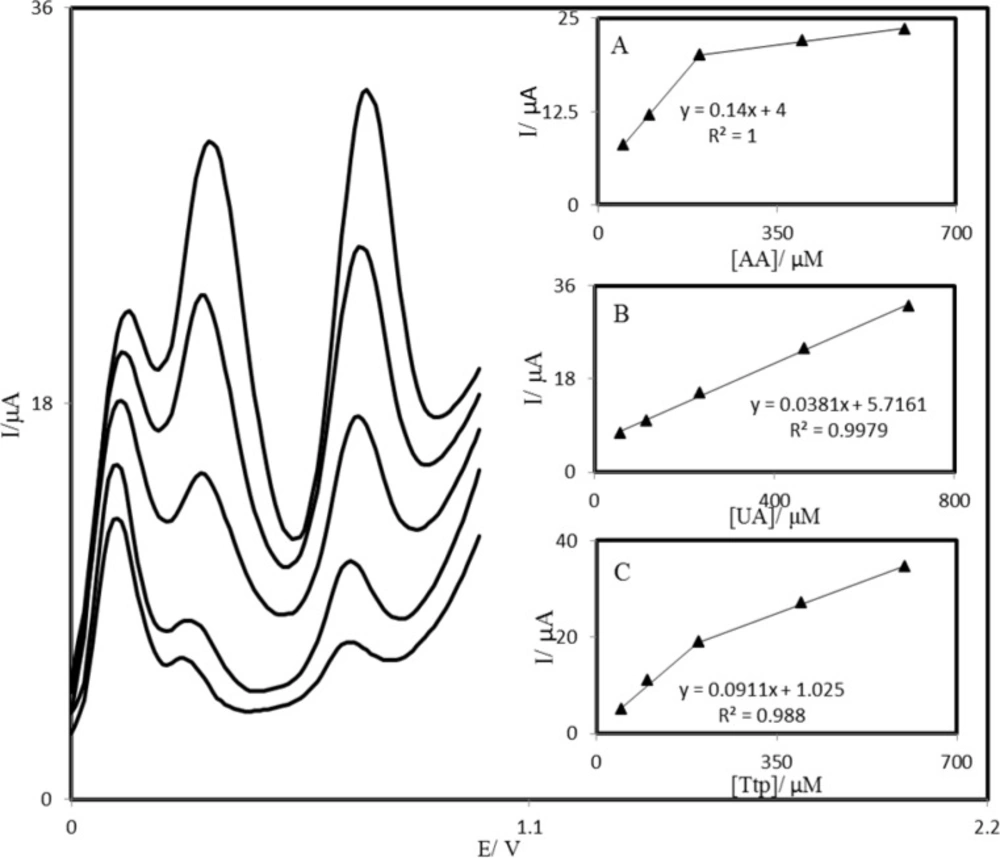
Simultaneous determination of AA, UA and Trp
The main objective of this study was to detect AA, UA, and Trp simultaneously. The utilization of the PBDCNPE for the simultaneous determination of AA, UA, and Trp was demonstrated by simultaneously changing the concentrations of AA, UA, and Trp. The AA voltammetric results showed that the simultaneous determination of AA, UA, and Trp with 3 well-distinguished anodic peaks at potentials of 100, 300, and 670 mV corresponding to the oxidation of AA, UA, and Trp, respectively, could be possible at the PBDCNPE (
Figure 9). The sensitivity of the modified electrode toward the oxidation of AA was found to be 0.143
μA
μM
−1, whereas the sensitivity toward AA in the absence of UA and Trp was found to be 0.147
μA
μM
−1. It is very interesting to note that the sensitivities of the modified electrode toward AA in the absence and presence of UA and Trp were virtually the same, which indicates the fact that the oxidation processes of AA, UA, and Trp at the PBDCNPE were independent; therefore, simultaneous or independent measurements of the 3 analytes are possible without any interference. If the AA signal were affected by the UA or Trp, the above-mentioned slopes would be different.
Interference study
The effect of a number of organic compounds such as uric acid, folic acid, captopril, cysteine, aspartic acid, tryptophan, glysine, acetaminophen and some ions such as chloride, potassium, nitrate, fluoride, sulfide, carbonate, and sodium on the determination of 1.0 × 10-4 M ascorbic acid was investigated.
The tolerance limit was taken as the maximum concentration of the foreign substances, which caused an approximately ±5% relative error in the determination. The tolerated concentration of foreign substances was 1.0 M for Na+, Cl-, F-, S2-, CO32-, HCO3-, NO3- and K+; 1.0 × 10-3 M for uric acid, folic acid, captopril, cysteine, aspartic acid, tryptophan, glysine and acetaminophen.
Real sample analysis
In order to evaluate the analytical applicability of the proposed method, it was applied to the determination of AA in vitamin C tablets, UA in urine and Trp in human serum samples. The DPV technique was used in the experiments. The differential pulse voltammograms were obtained by spiking appropriate samples in diluted solution using PBDCNPE at optimum conditions as described earlier. The results for determination of the three species in real sample are given in
Table 1. From these results, it can be seen the PBDCNPE shows good activity for real samples analysis. This procedure was repeated five times and the relative standard deviation were 1.8%, 2.1% and 2.5% for AA, UA and Trp, respectively.
The repeatability and stability of PBDCNPE
The ability to generate a reproducible electrode surface was examined using cyclic voltammetric data from five separately prepared PBDCNPEs, obtained in optimum solution pH. The calculated RSD for various parameters (2–4%) indicated that surface reproducibility was satisfactory. This degree of reproducibility is virtually the same as that expected for an ordinary carbon paste surface (43). In addition, the long-term stability of the PBDCNPE was tested over a three-week period. When CVs were recorded after the modified electrode was stored in atmosphere at room temperature, the peak potential for AA oxidation was unchanged and the current signals showed less than 2.3% decrease relative to the initial response. The antifouling properties of the modified electrode toward AA oxidation and its oxidation products were investigated by recording the cyclic voltammograms of the modified electrode before and after use in the presence of AA. Cyclic voltammograms were recorded in the presence of AA after having cycled the potential 10 times at a scan rate of 25mV s-1. The peak potentials were unchanged and the currents decreased by less than 2.5%. Therefore, at the surface of PBDCNPE, not only does the sensitivity increase, but the fouling effect of the analyte and its oxidation product also decreases. The surface of the PBDCNPE was regenerated before each experiment.