Materials
Chitosan, Glycine, Sodium borohydride (NaBH4), Penicillin/ Streptomycin (Pen Strep), Myristic acid, EDC [1-ethyl-3 (3-dimethylaminopropyl)Carbodiimide], NHS (N-hydroxysuccinimide), MTT[3-(4,5-dimethylthiazol-2-yl) -2,5-diphenyltetrazolium bromide] and Rhodamin 123 were from Sigma Aldrich (USA). Glacial acetic acid (100%), DMSO (Dimethyl Sulfoxide), BSA (Bovine serum albumin) were from Merck (Germany). Doxorubicin hydrochloride (DOX-HCl) was from EBEWE PharmaGes (Austria). Agarose and Low Melting Agarose were from Banglor Genei India Co. RPMI 1640 medium and fetal bovine serum (FBS) were from Gibco (Invitrogen). The LNCaP and PC-3 cell lines were from the Pasteur Institute of IRAN. All other chemicals used in this research were of analytical grade from Merck (Germany) or Sigma Aldrich (USA).
Buffers
DPBS (pH 7.2-7.4): CaCl2.2H2O 0.1 g, Glucose 0.5 g, KCl 0.2 g, KH2PO4 0.1 g, MgSO4.7H2O 0.1 g, Na2HPO4 0.05 g and NaCl 4 g.
Wash buffer (WB): DPBS 500 mL, Glucose 2.25 g and MgCl2 1M 2.5 mL.
Binding Buffer (BB): DPBS 500 mL, Glucose 2.25g, MgCl2 1M 2.5 mL and BSA. Keep in 4 °C for 1 month.
Cell Culture
The LNCaP cells as a positive target and PC-3 cells as a negative control were cultured in RPMI 1640 under standard cell culture conditions supplemented with 10% FBS and 1% Pen Strep/ 37 oC in a humid environment with 5% CO2.
Primers and Library
The forward primer with the following sequence was labeled at the 5′-end with –NH2 for conjugation of aptamer to nanogel.
5`-NH2-CATCCATGGGAATTCGTCGA 3`
The reverse primer with the following sequence was labeled at the 5′ end with phosphate for selective digestion of the phosphorylated strand of double stranded DNA (dsDNA) from the 5’ end of PCR products by an exodeoxyribonuclease enzyme (Lambda exonuclease) as described by Marimuthu (
13).
5`-p- CTGCCTAGGCTCGAGCTCG 3`
The initial library with the following sequence were designed and obtained from the GSFLX Company (German).
5` CATCCATGGGAATTCGTCGAC (N) 40 CGAGCTCGAGCCTAGGCAG 3`
Cell-SELEX Procedure
Method of Kang (
16) with some modifications was used for Cell-SELEX. In brief, in each round of selection, 5 nmol of ssDNA aptamers per 1mL BB, was first folded by incubation/ 95
oC for 10 min and immediately cooled on ice for 15 min. ssDNA aptamers was incubated with approximately 10
6 live washed LNCaP cells with gentle shaking/ room temperature (RT). After 1 h, the cells containing the binding aptamers were collected with centrifugation/ 1000 RPM for 3 min/ 4
oC and washed with WB. The pellet was heated/ 95
oC for 5 min in 500 µL DNAase-free water and the supernatant containing aptamers were collected and amplified by PCR. The PCR product was treated with Lambda exonuclease and the concentration of ssDNA was determined by a nanodrop. Five nmol of the ssDNA was used for the second round of SELEX. The steps were repeated for 10 rounds of SELEX with gradually increasing the stringency of the reaction by decreasing the incubation time from 1 h to 15 min and increasing washing buffer volume from 1 mL to 10 mL.
In order to eliminate the nonspecific binding aptamers to common antigens, the counter SELEX was carried out with the incubation of the DNA aptamers obtained from the 3rd and 7th rounds of SELEX with 106 live PC-3 cells. The cells were precipitated and the supernatant containing the unbounded aptamers was amplified and used in further SELEXes.
Cloning and Sequencing of Enriched Pools
Enriched aptamers with high affinity to LNCaP cells were collected, PCR amplified and were cloned into the pTG19-T plasmid and was transformed into E.coli. Positive clones resulting from transformation were selected and the affinity of the candidate aptamers to LNCaP cells was assayed separately. Two of the clones with highest affinity were sequenced.
The binding affinity of the finally selected aptamer was estimated by flow cytometry. Target cells (1 × 106) were added to varying concentrations of FITC-labeled aptamers (0, 50, 100, 150, 200 pmol) in a 200 μL volume of BB. The original library was used as a negative control. The dissociation constant (Kd) was estimated by the equation Y = B max X/ (Kd + X), using Sigma Plot 12.0 software (Jandel, San Rafael, CA).
Preparation of Myristylated Chitosan (MCS) Nanoparticles
A homogeneous solution was prepared by dissolving 0.250 g Chitosan powder in 50 mL acetic acid aqueous solution (2%) and stirred for 20 min/room temperature (RT). The 0.5% (w/v) solution of CS was sonicated with the power of 70W for 20 min/18
oC. CS nanogels was cross linked with Myristate using the EDC / NHS technique described by Cheng
et al. (
18). CS nanogels (5 mg/mL) was mixed with 2 mL solution of EDC (90 mg) and NHS (55 mg) and incubated at RT for 5 min. 0.036 g Myristate was dissolved in ethanol and added drop wise to the above mixture with continuous stirring for 30 min/RT to obtain a Myristate: CS ratio of 1:9. The mixture was added with ethanol and stirred continuously for another 5 h. The solution was precipitated by increasing the pH to 10 with a 10 M NaOH solution and then centrifuged for 20 min at 5000 g /18
oC. In order to remove the unreacted Myristate, EDC and NHS residuals, the pellet was washed with ethanol followed with distilled water (DW). The precipitated gel was dissolved completely in 2% acetic acid solution and labeled as S1. To find the best storage condition, one mL of S1 nanogel was freeze-dried and stored at RT as S2 sample. Whenever needed, the S2 sample was dissolved in 1 mL of 2% acetic acid with sonication.
Nanogel Characterization
Particle Size Measurement
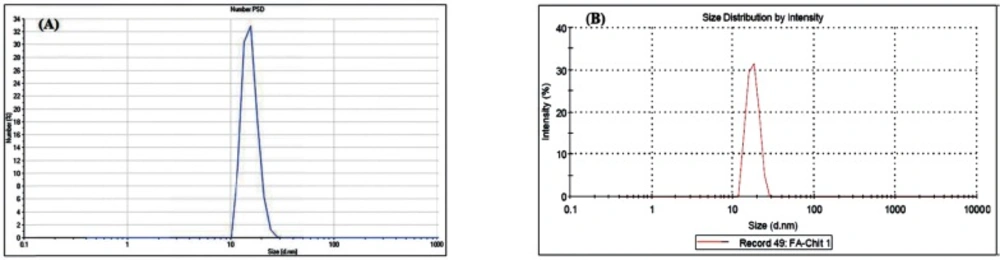
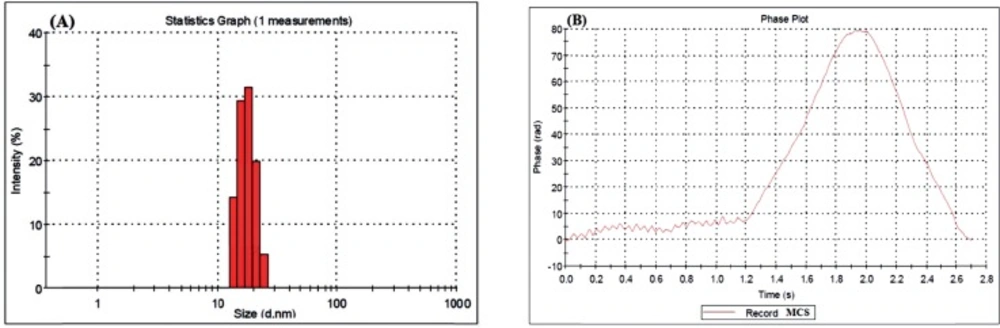
The nanoparticles were sputter-coated with a thin layer of gold and scanned by KYKY-EM3200 Scanning Electron Microscopy (SEM) at 25 KV accelerating voltages. The mean particle size and particle size distributions of the nanogels were also determined by Zeta plus Dynamic Light Scattering (DLS) Zeta Sizer Nano-ZS-90 (Malvern Instruments). The mean particle size was measured for 120 replicates and Poly dispersity Index (PdI) were calculated. To disperse the nanoparticles, samples were sonicated in a water bath. The Zeta Plus instrument was used to measure the electrophoretic mobility of the MCS nanogels.
FT-IR Test
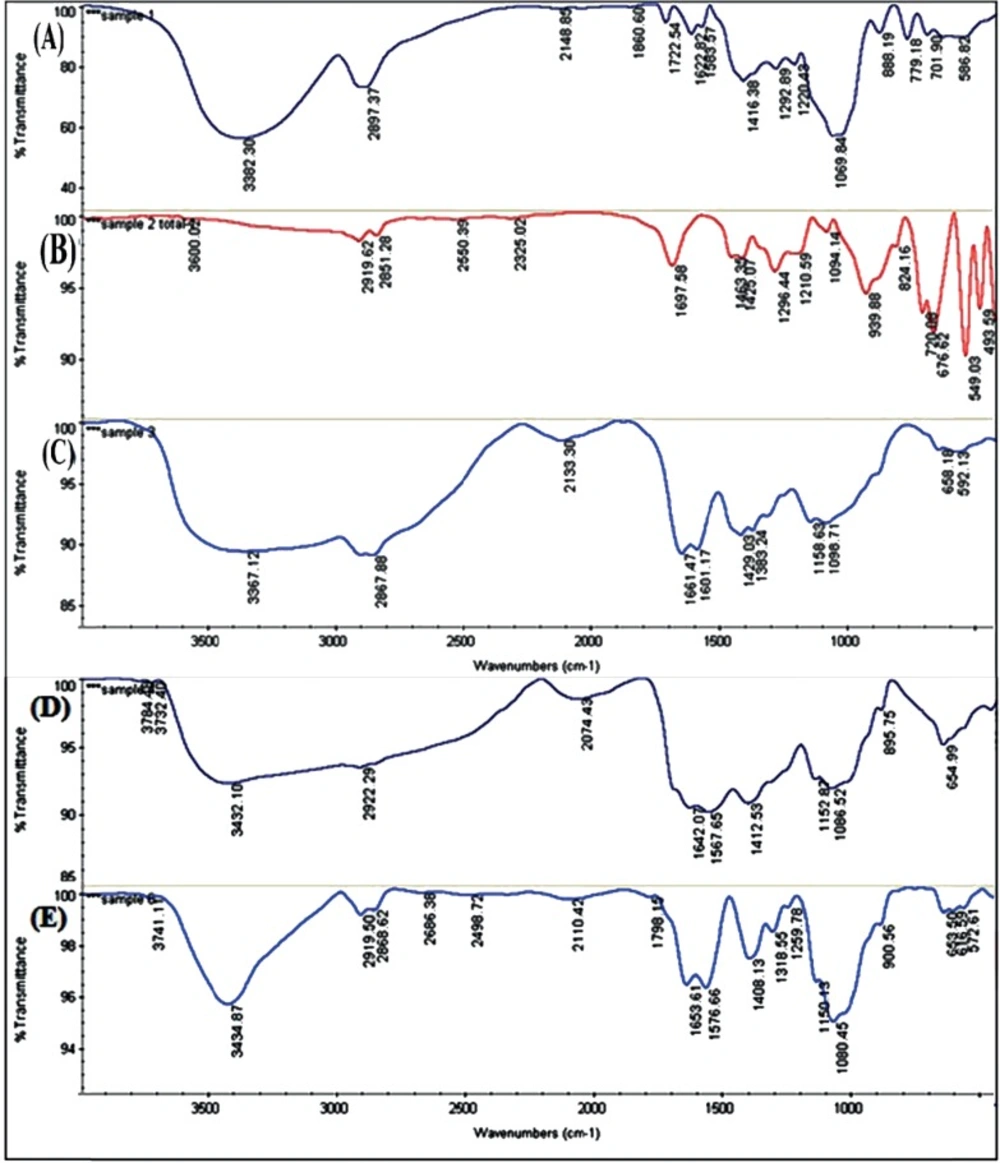
Spectral analyses of CS, Myristate, MCS, DOX and MCS-DOX carried out by Fourier-transform infrared spectroscopy (FT-IR) using Thermo Nicolet Nexus 870 FT-IR ESP (USA) spectrophotometer. Approximately 100 mg of each of the above dry samples was grounded with KBr and converted to tablets under a pressure of 600 Kg/cm2 by hydraulic press and used for FT-IR analysis.
Drug Loading and Release
Method of Wang
et al. (
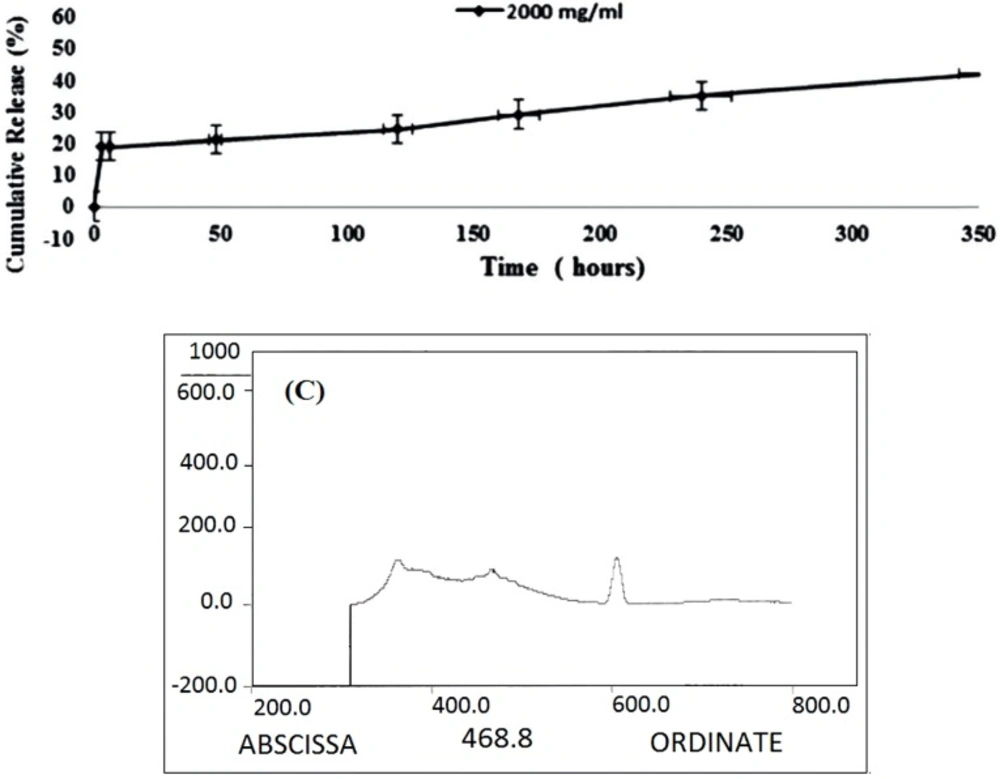
19) was used to estimate the drug loading and release capacity of the gel. 5.0 mg/mL of MCS nanogel was incubated with 2 mg/mL of DOX. The solution was mixed thoroughly for 24 h/4 °C under shaking condition. The excess free DOX was removed by dialysis using 2 KDa MWCO dialysis bags against 30 mL DW. The DOX loaded MCS was then dialyzed against 30 mL of phosphate-buffered saline, pH 7.4/ 37 °C. At various time intervals 2 mL of phosphate-buffered saline from each dialysis buffer was taken and analyzed by HPLC for the presence of DOX. The buffer taken for analysis was replaced with an equal volume of fresh buffer.
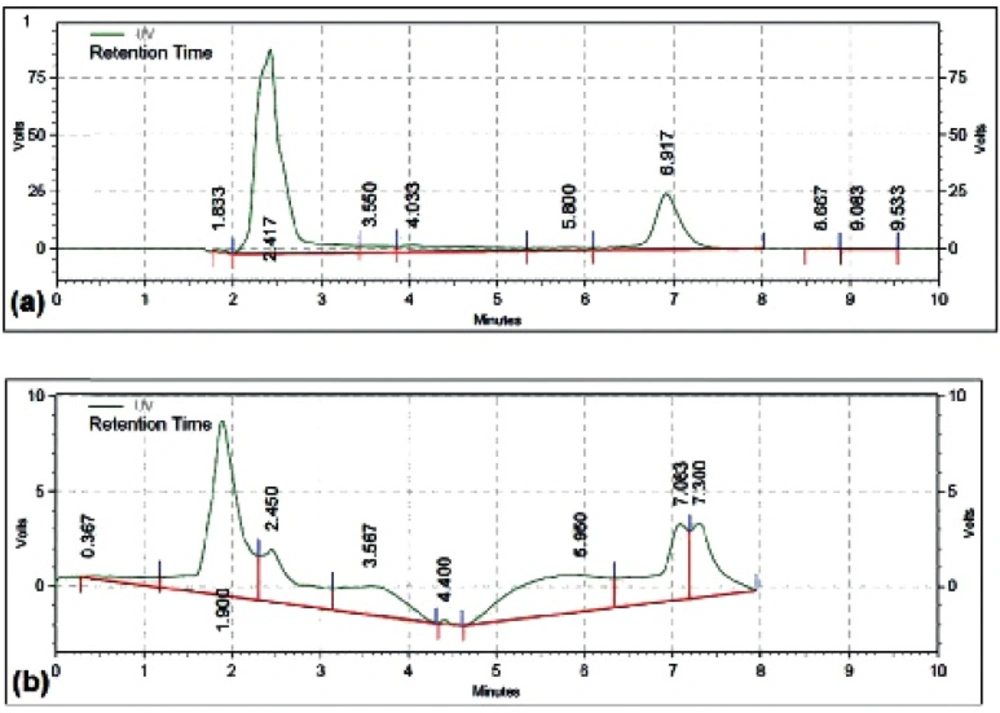
Determination of Loading Capacity and Release Rate of DOX
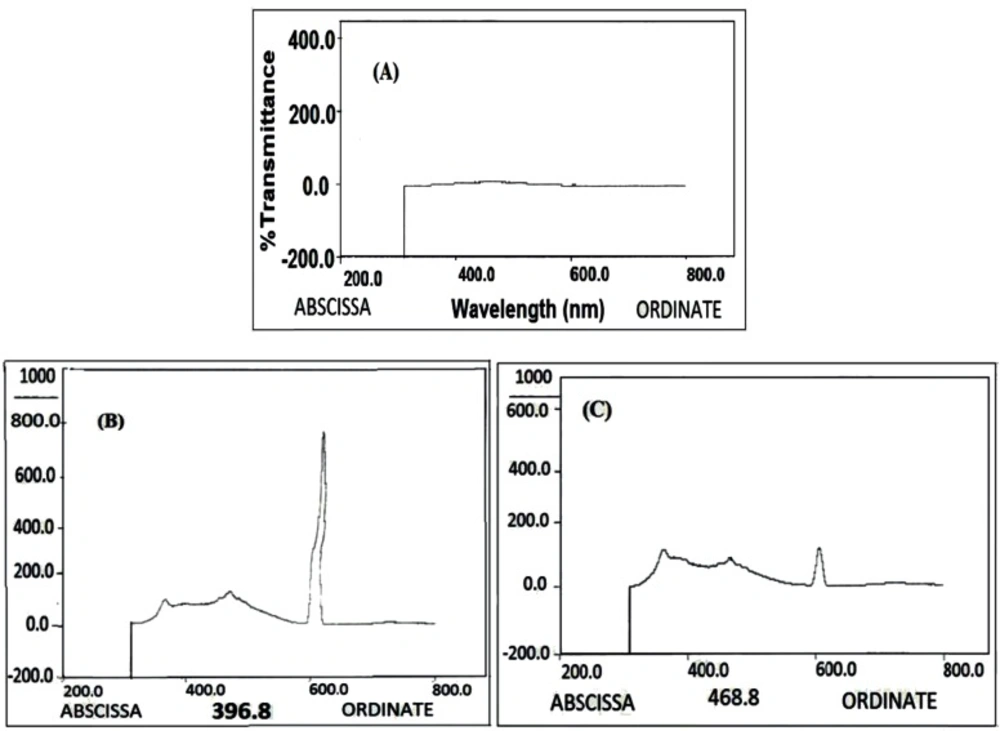
The amounts of the loaded and released DOX were determined by High-performance Liquid Chromatography (HPLC) using a reversed phase column (C18, 5 μM, 250 mm× 4.6 mm).
The concentration of DOX was calculated using a standard curve of DOX. Different concentrations of DOX were injected into the column and eluted with sodium lauryl sulfate-Phosphoric acid: acetonitrile of 40:60 ratio with a flow rate of 1 mL/min. The sample solutions were detected with a run time of 7 min for assay.
The OD of DOX was read at 254 nm. Following equations was used to calculate the loading efficiency (LE) and loading capacity (LC) of the DOX:
LE = (Total amount of DOX − Free DOX) / Total amount of DOX
LC = (Total amount of DOX − Free DOX) / amount of MCS
Synthesis of MCS-GA-CHO Polymer
Aldehyde-functionalized polymer MCS-GA was synthesized by conjugating
CHO-GA-CHO to MCS-NH2 by the drop wise addition of 0.03 mM MCS nanogel to 15.6 mM GA to obtain the 1:500 ratio. The Schiff base cross-linking involves a reaction between the amine groups of Chitosan and the -CHO groups of glutaraldehyde. The yellow color developed indicates the reaction progress. The excess GA was removed by dialysis of the mixture against 1 L DW/ RT for 24 h using 3 KDa MWCO dialysis bags. The MCS-GA was stored at 4 oC after sonication.
Conjugation of Aminated ssDNA Aptamers to MCS-GA-CHO Nanogels
To attach the DNA aptamers to MCS-GA-CHO nanogels using glutaraldehyde as a linker, the aptamers were amplified with the NH2–labeled forward and the phosphate-labeled reverse primers. Aminated DNA aptamers were treating with lambda exonuclease in order to get ssDNA. The ssDNA was denatured/ 90 oC for 10 min and then incubated on ice for 15 min to obtain their native folding. 2-4 nM of ssDNA aptamers was added drop wise to 1 mg MCS-GA-CHO nanogels in the total volume of 1mL and incubated for 6 h under shaking condition. In order to monitor the MCS-GA-Apt nanogels, 2 µL of 5 nM Rd was added to the mixture. After 1h shaking, 5 mg Glycine dissolved in 0.5 mL DW was added to the mixture and shacked for another 30 min. 15mg NaBH4 dissolved in 1 mL DW was added drop wise to the mixture with continuous shaking for 30 min. The mixture was centrifuged at 12000 g/ 10 min and washed twice with DW and resuspended in BB by sonication and stored/ 4 oC.
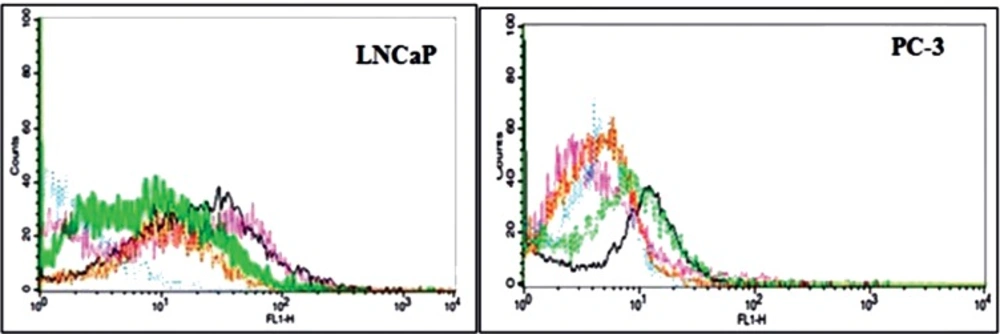
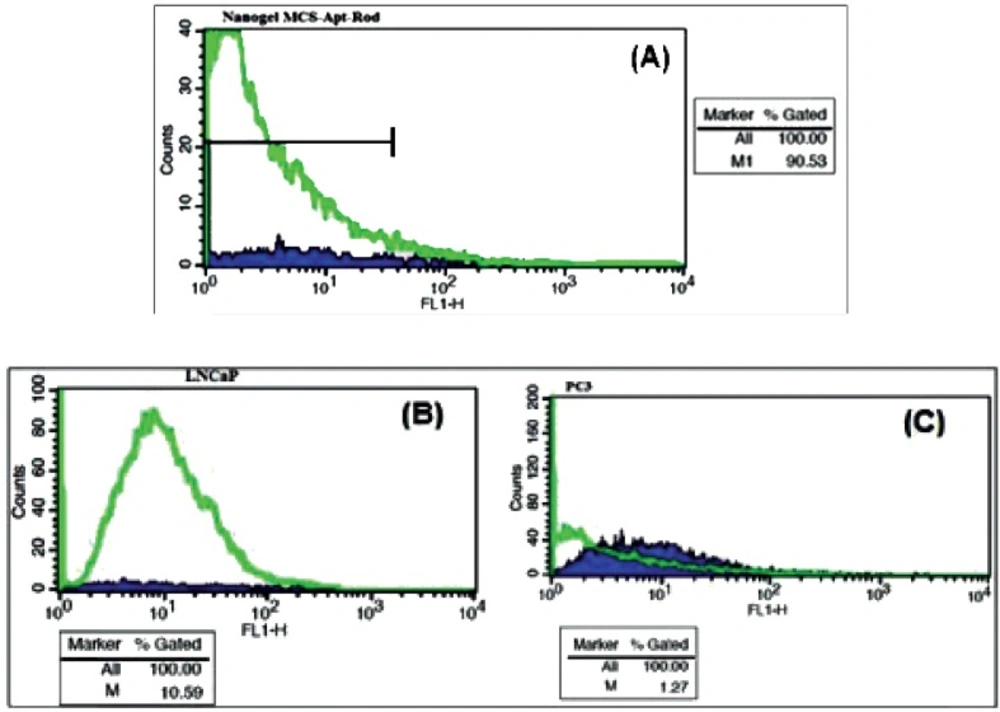
Flow Cytometry Binding Assays
The affinity of the aptamer conjugated nanogel to the Pcanc cells was evaluated by Flow cytometry. Approximately 106 live LNCaP and PC-3 cells were washed and resuspend in 100 μL BB. 1 mL MCS-Apt/Rd in BB was incubated with both LNCaP and PC-3 cells for 1 h/ 37 oC in a humid environment with 5% CO2. 50 μL from each cell lines were diluted in 1 mL BB and was pipetted into different flow cytometry tubes. The fluorescence intensity of Rhodamin of each tube was determined against the untreated cells alone as a background.
DOX-Loading in MCS-GA-Apt Nanogels
10 mg DOX-HCl powder was dissolved in 1 mL DMSO. 0.5 mL DOX in DMSO was mixed with the MCS-GA-Apt nanogels and incubated for 2 days/4 oC under shaking condition. The mixture was dialyzed against DW using a dialysis tubing with a cutoff of 3 KDa for 24 h to remove the nontrapped DOX.
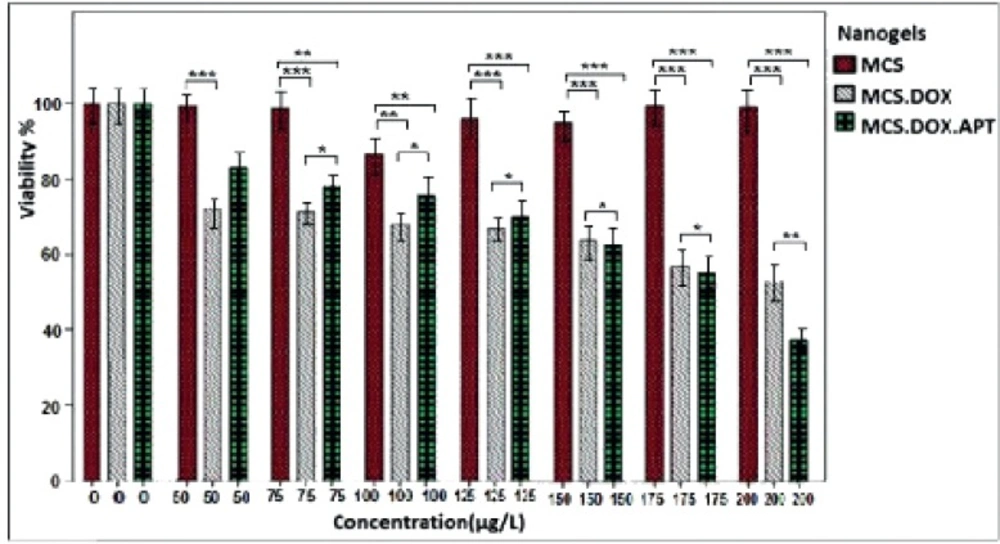
In-vitro Cell Viability
The relative number of the viable cells after treatment was determined by MTT assay. 1×104 LNCaP cells/well were plated in 96-well plates containing RPMI medium, 10% fetal bovine serum and 1% Pen Strep. After 24 h, the medium was replaced with various concentrations of DOX in RPMI medium containing 1% Pen Strepwithout FBS. After 24 h, 20 μL of MTT (3 mg/mL) was added to each well and the plates were incubated/ 37 °C for 5 h. The cells were then centrifuged at 700 g for 15 min and the supernatant was discarded. The cell pellets were dissolved in 100 μL of DMSO and the OD570 was measured. The untreated cells were considered as a control. The percentage of viability was determined by the following formula:
%Cytotoxicity = 1- (mean absorbance of toxicant / mean absorbance of negative control) × 100
%Viability = 1- % Cytotoxicity
The MTT assay was repeated with 0-200 µM of each of MCS, DOX-loaded MCS (non-targeted) and MCS-GA-Apt (targeted) nanogels and the percentage of viability was determined as above.
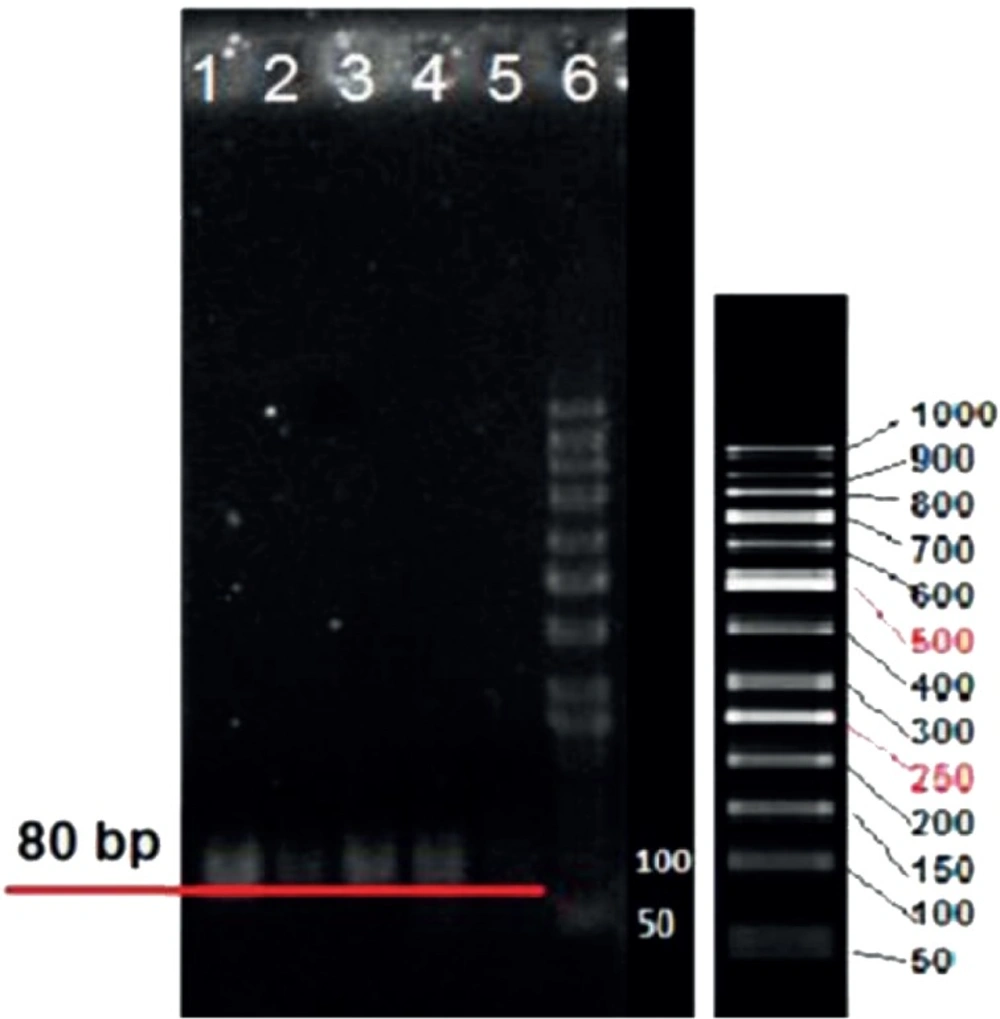
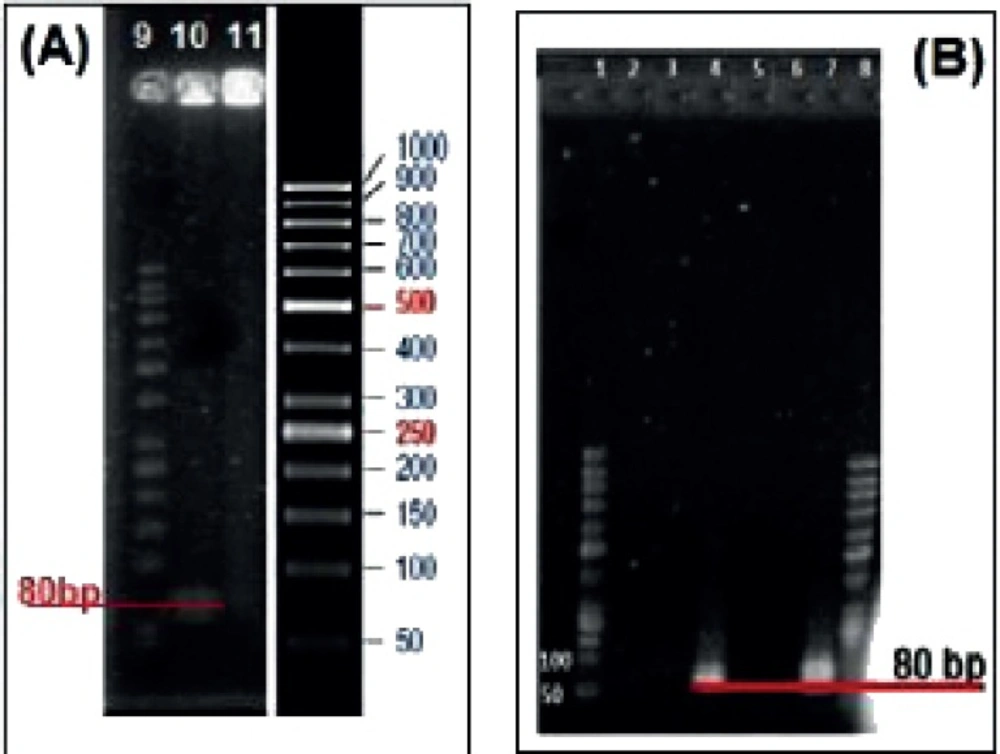
Gel Electrophoresis
To confirm aptamer conjugation, MCS-GA-Apt nanogels were loaded into the 1% agarose gel along with the molecular weight ladder. The electrophoresis was performed at 50V and the gel was stained with 0.5 mg/mL Et. Bromide.
Statistical Analysis
The statistical differences between the test and control samples were determined by Analysis of Variance: ANOVA One-Way with P < 0.05. Values are considered as means ± standard deviation for at least three independent experiments by Tukey analysis.