MMP-1(human fibroblast collagenase) is secreted by stromal fibroblasts, osteoblasts, and osteoclasts. It cleaves native collagen (abundant in bone matrix) at neutral pH (
1). Overexpression of this enzyme is usually associated with the irreversible degradation of cartilage, tendon, and bone in people with arthritis (
2).
Various types of mammalian collagenolytic enzymes are involved in the degradation of extracellular matrix including matrix metalloproteinases (MMPs), cathepsin K, and neutrophil elastase (
3). The MMPs are members of a family of zinc-dependent proteolytic enzymes. They mostly appear key players in endochondral ossification during embryonic development, modeling, remodeling and homeostasis of the skeletal system (
4,
5).
A cumulative evidence supports that MMP-1 cleavage activity is involved in many biological process such as keratinocyte migration and reepithelization, platelet aggregation, increased bioavailability of insulin-like growth factor 1 (IGF-1), cell proliferation, protease activated receptor-1 (PAR1) activation and pro-inflammatory and anti-inflammatory processes (for review, see reference 6 ). MMP-1 is also upregulated in tumor tissues and has been suggested to be associated with tumor invasion and metastasis (
7). There is also a unifying link between MMP-1 and lung cancer risk (
8). The MMP-1 significant contributions to these diverse biological and pathological conditions denote the probable involvement of this metalloproteinase in the pathogenesis of cancer and autoimmune diseases such as rheumatoid arthritis.
The active enzyme form of MMP-1 contains zinc- and calcium-binding catalytic domains with a COOH-terminal which may be involved in matrix binding. The structure is consisted of a twisted five-stranded beta sheet and three long helices. The collagenase active site cleft is bordered by a beta strand, a helix and a stretch of random coil adjacent to the COOH-terminus of that helix. The catalytic zinc is at the bottom of the cleft and is ligated by three conserved histidine residues, each at a distance of 2.1 Å. The second zinc ion is coordinated by two His and one Asp in an approximately tetrahedral manner. The calcium ion presented in the enzyme is ligated in an octahedral manner by two Asp and one Glu and two Gly and one Asn. Earlier studies demonstrated that zinc and calcium significantly stabilize the tertiary structure of collagenase (
9).
Inhibition of MMP-1 activity may affect bone remodeling under certain pathological conditions. This can be related to the upregulation of interstitial metalloproteinases and bone loss. In order to selectively inhibit MMP-1 for therapeutic intervention, combined sophisticated theoretical and experimental approaches are required (
10).
Studies on MMP inhibition, principally the structural determination of enzyme-inhibitor complexes, are crucial for understanding the structural features associated with inhibition. This information includes the scientific basis for a rational search for new and more potent inhibitors, through theoretical chemistry calculations (
11,
12,
13).
Traditionally, the design of an MMP inhibitor (MMPi) has involved the use of zinc-binding groups due to the fact that MMP cleavage on substrates directly involves the catalytic Zn
2+ ion(s) (
14,
15,
16). Upon binding of the substrate, the zinc-bound water molecule attacks the substrate carbonyl carbon atom and the transfer of protons through a preserved Glu residue to the amide nitrogen of the scissile bond results in peptide cleavage (
9). Therefore, zinc-binding groups (ZBG) in MMPi displace the zinc-bound water molecule and inactivate the enzyme (
11). In addition, the ZBG acts as an anchor to lock the MMPi in the active site and directs the inhibitor into the target substrate-binding pockets.
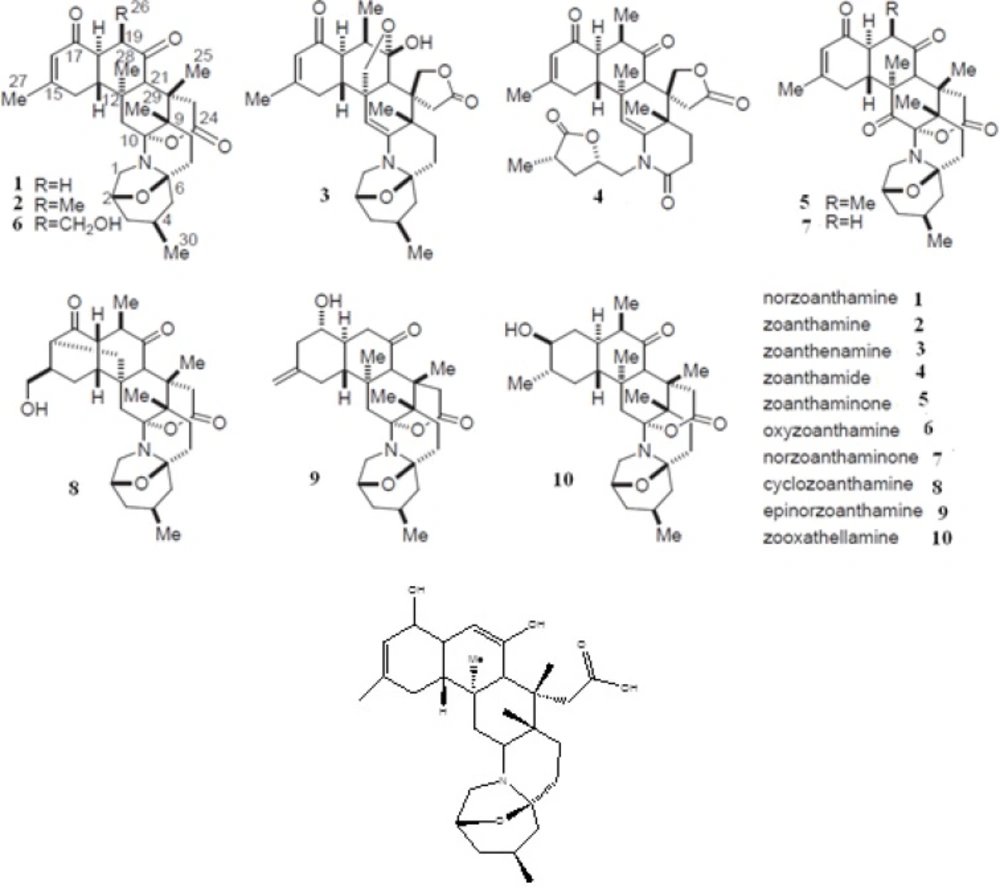
As one of the members of zoanthamine class of marine alkaloids, there exist high concentrations of norzoanthamine in the epidermal tissues of
Zoanthussp (
17). Over the past 20 years, more than twelve nitrogenous marine metabolites known as zoanthamine alkaloids have been reported. They show a wide spectrum of biological effects including the inhibition of phorbolmyristate acetate-induced inflammation in mouse ear (
18); inhibition of the growth of P-388 murine leukemia cell lines (
19); inhibitory effects on thrombin-, collagen-, and arachidonic acid-induced human platelet aggregation (
20); suppressive effects on interleukin-6 production (
21); anti-osteoporosis activity in ovariectomized mice (
22); acceleration in the formation of the type I collagen-hydroxyapatite composites (
23); enhancement of collagen release from the immobilized matrix vesicle model and suppression of the proteolysis of bovine serum albumin, elastin and collagen (
23).
Moreover, it has been suggested that collagen-norzoanthamine supramolecular association may be the mode of anti-osteoporosis action of this marine alkaloid (
23). The collagen protective activity of norzoanthamine suggests that these marine alkaloids may be a promising drug candidate for the prevention or treatment of osteoporosis-associated bone loss. In the present study, we examined the inhibitory effects of norzoanthamine and its homologues on human fibroblast collagenase by using theoretical chemistry studies.
Methods and Computational Details
The crystal structure of fibroblast collagenase was extracted from PDB bank (PDB code: 1CGL) (
9). This structure refers to collagenase co-crystallized with an inhibitor (N-[(1s)-3-{[(benzyloxy) carbonyl] amino}-1-carboxypropyl]-l-leucyl-N-(2-morpholin-4-ylethyl)-l-phenylalaninamide) (OED). Since PDB structure has two identical chains and collagenase has only one chain, the chain A of protein was just retained for this study. 2D structures of the studied ligands (molecules 1 to 10 and the molecule1 in its enol-imine tautomer known as ligand 1-enol), were drawn (
Figure1) and minimized via Hyperchem 8 software (
24). They were then optimized at the density functional theory (DFT) level. The B3LYP hybrid functional was applied to these systems (
25,
26). For electronic and structural investigations of ligands, the 6-31+G* basis set was considered. Finally, the vibrational frequencies were computed to elucidate the global minimum structure. All calculations were performed using the Gaussian 2003 program (
27). The binding affinities between the ligands and the active sites of the enzyme were studied using docking studies (AutoDock 4.2 suites,
28). MD simulation and MM-PB/GBSA binding free energy calculations were performed via Amber package version 12 (
29). All calculations were repeated with a crystallographic ligand (OED) as control. Afterwards, eleven ligands were studied.
Molecular docking
All ligands plus the co-crystallized inhibitor (OED) were docked into the chain A of original PDB file. The estimated binding affinities between the ligands and the active sites of the enzyme were then explored by docking studies (AutoDock 4.2 suites) (
28). Input files required for AutoDock 4.2 were made in Autodock Tools (ADT) (Autodock Tools, 3.0). The Gasteiger charges were assigned to the ligands. All files were converted to the PDBQT format needed for Autodock calculations. The protein and ligand were considered rigid and flexible, respectively during docking procedure.
At first, docking calculations were carried out with performing a blind docking. The grid box was centered on the original ligand position of the crystal structure (OED). Then the lowest docking energy of each ligand was selected. Further docking calculations were done to obtain the top-ranked pose. All hydrogen atoms were added to the collagenase protein and after assigning Kollman united atom charges and atomic solvation parameters, then the non-polar hydrogens merged. The size of the grid box for final docking to specify the search space was set at 60×60×60, and centered on the predicted cavities with a default grid point spacing of 0.375 Å. Pre-calculated grid maps storing interaction energy of the ligand atom type with the receptor target, were obtained using AutoGrid4.
The zinc parameters used in this study were radius(r = 1.1Å) and well depth (ε = 0.25 Kcal/mol) (
31). A charge of +2.0 e was assigned to the zinc and calcium ions (
31) in PDBQT file.
The Lamarckian Genetic Algorithm (LGA) (
28) was used for a conformational search approach with 200 runs of Genetic Algorithm runs. A maximum number of 2,500,000 energy evaluations per run were carried out, while crossover and mutation rates were run with the default parameter settings. The step size parameters were changed, i.e. "Translation" to 0.2 Å, "Quaternion" to 5.0°, and "Torsion" to 5.0°.
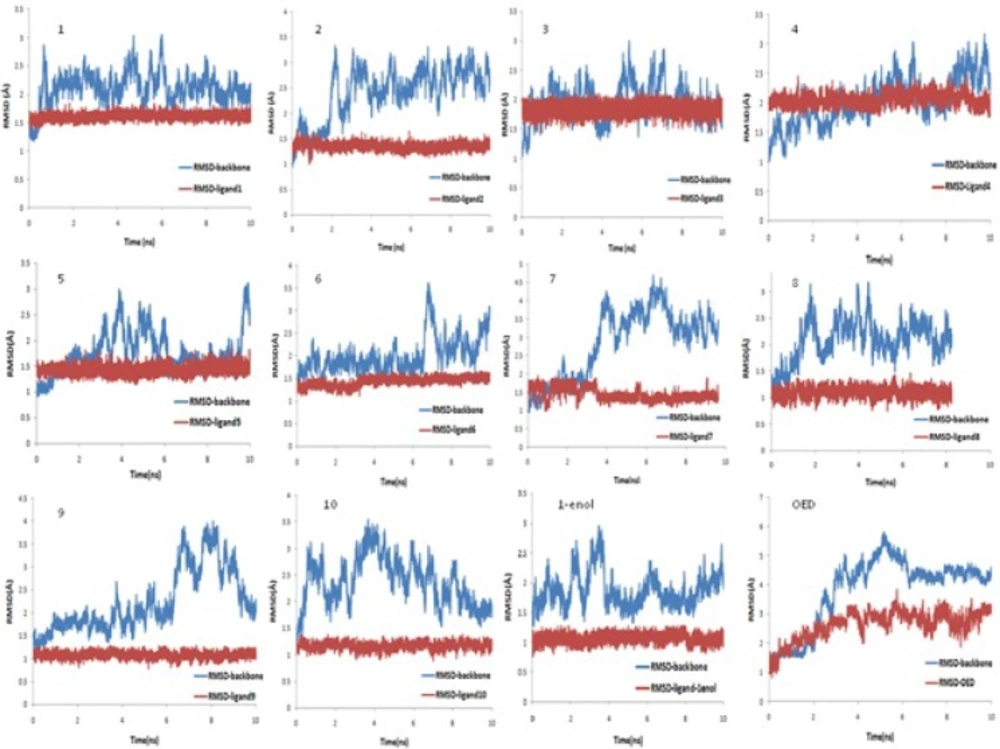
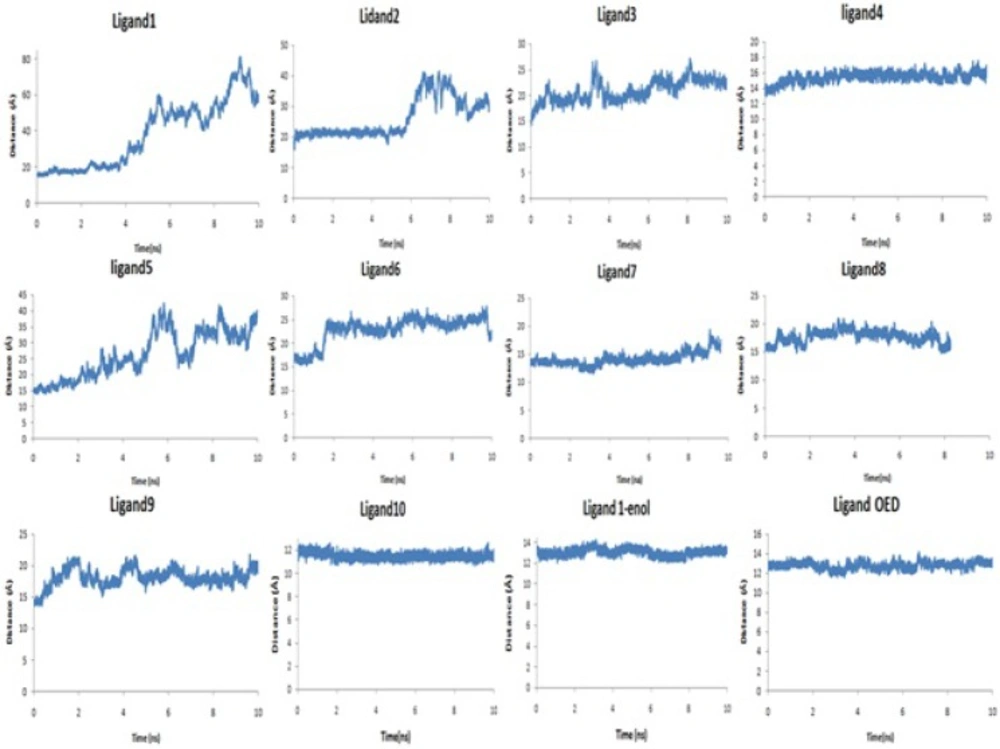
AutoDock4.2 applied to evaluate the binding free energy was used to discover the best binding mode. The energy scoring values of AutoDock4.2 are consisted of intermolecular energy (van der Waals, hydrogen bonding, desolvation and electrostatic energies), total internal energy and torsional free energy. In fact the lower the binding free energies, the more effective the ligand-binding protein. The root-mean-square deviations of ligands (RMSD) were calculated in order to put them into families of similar conformations or "clusters". Therefore, the expected docked complexes are preferred based on the lowest binding free energy from the largest"clusters" for each ligand.
The docked conformations of each ligand were ranked into clusters based on the binding energy. The results were clustered with the tolerance of 2.0 Å RMSD. Based on the lowest binding free energy of the most populated cluster, the best position of each ligand was chosen for subsequent studies.
Molecular dynamics (MD) simulations
The best docked complexes were selected for MD simulations. Twelve MD simulations (11 ligand and OED) were performed using the Amber12 package (
29). The protein was modeled using the AMBER FF99SB force field. The inhibitor charges were obtained using the RESP fitting procedures (
32). Topology prep files for ligands were built with the general amber force field (gaff) using Antechamber module in AmberTools12 (
33). The complexes were then solvated in a truncated octahedral box of TIP3P (
34) water molecules with a buffer size of 12 Å, and neutralized using 10 Na
+ ions. The default protonation of Amber12 was used for titratable residues in protein.The energy minimization and MD simulations were performed for each system using the SANDER module of the Amber12 package (
29).First, 10000 cycles of minimization (MAXCYC) with the first 700 steps of steepest descent (NCYC) and 9300 steps of conjugate gradients on all atoms of the system were performed to relieve the bad steric interactions and to get closer to an energy minimum. After energy minimization, the position restraints at constant volume (NVT) for 100 ps, with a restraint force of 10 Kcal/mol at temperature of 100 K, and then at constant pressure (NPT) for 100 ps, with a restraint force of 1 Kcal/mol at temperature of 300 K, were performed.
Next, an equilibration step at constant pressure (NPT) at 300 K was performed. The duration time in this step was 100 ps and the time step was 2 fs, while the restraint force was removed in this step. This approach allows the water to equilibrate around the protein and come to an equilibrium density. In the position restraints and equilibration steps, Langevin dynamics were used to control temperature.
Finally, a production step for 10 ns with a 2 fs time step at constant pressure (NPT ensemble) and with an isotropic position scaling (ntp = 1) at 300 K was carried out. In this step, the Berendsen thermostat was used to control temperature (ntt = 1). All bonds with hydrogen atoms in all steps were constrained via the SHAKE algorithm (35) (ntc = 2, ntf = 2). The periodic boundary was used in all steps (ntb = 1). The long-range electrostatic interactions were treated using the particle mesh Ewald method (
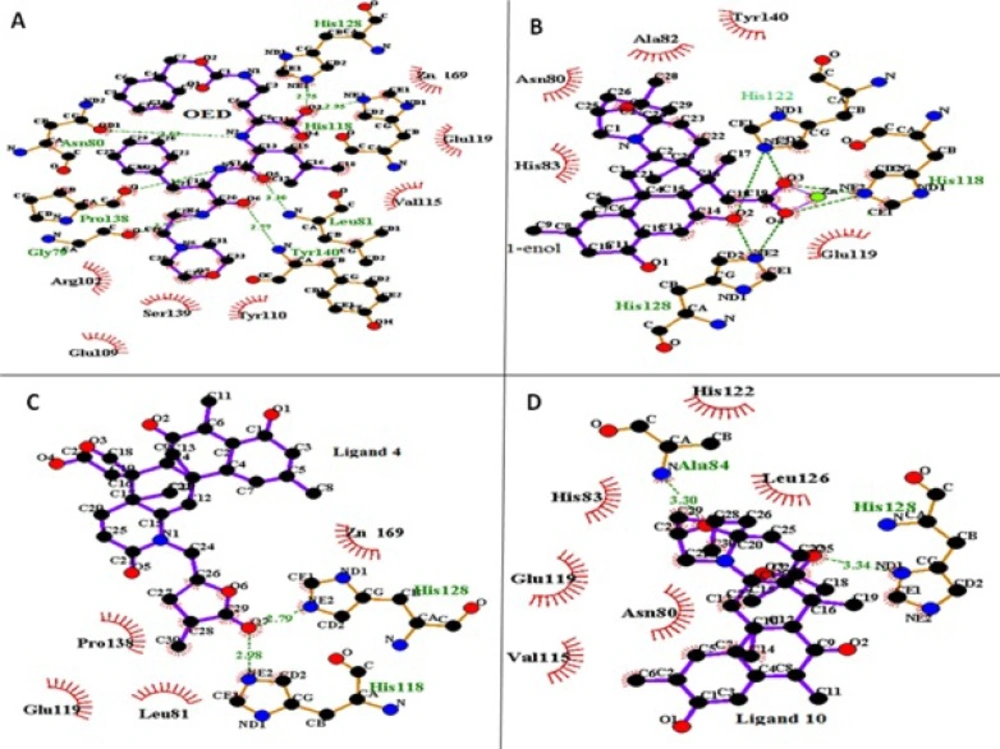
36), and a nonbonded cutoff was set to 10 Å. The zinc and calcium ions were connected to related residues via tleap module of Amber12. The images of ligands in the active site of protein were generated with LigPlus (
37).
MM-PB/GBSA calculations
Molecular mechanics Poisson Boltzmann/surface area (MM-PBSA) is regarded as an attractive approach for drug design since it works not only for small and medium size organic compounds but also for the biological molecules such as proteins and DNA. The MM/PBSA is a molecular dynamics based approach that is fast and practical for binding free energy calculations, in spite of free energy perturbation (FEP) or thermodynamics integration (TI) that are time-consuming. Also, unlike the linear interaction energy (LIE) method, MM-PBSA applies no empirical parameters in its free energy calculations. It does not need a training set to derive the empirical parameters as does LIE, which makes it a promising method for ranking very different compounds from database searching for binding to a given site (
38). Therefore, by combining this method with molecular docking and molecular dynamics simulations, one can hope to reliably model a protein and DNA complex (
38).
The binding energies in this study were calculated by the MM-PBSA method as implemented in Amber 12 (
39-
43). Four hundred snapshots were extracted from MD simulation trajectories for binding free energy calculations. The water molecules and counter-ions were stripped.
In summary, the binding free energy is:
Each free energy term in Eq (1) is computed using Eq(2):
The entropy term is written as T∆S. ∆Egas stands for the gas phase or vacuum molecular mechanical energy and can be divided into the van der Waals, electrostatic, and the internal energy contributions in gas phase. ∆Gsolvation is the solvation free energy and estimated using continuum solvent methods. It can be partitioned into two parts: the polar contribution (∆G(PB/GB)) and the nonpolar contribution (∆Gnonpolar) as shown in Eq(3).
The electrostatic solvation free energy was determined using the finite difference PB or GB models. The nonpolar contribution to the solvation free energy was calculated from the solvent accessible surface-area.
Where ∆SASA (solvent accessible surface-area) was determined with linear combinations of pairwise overlaps (LCPO) method (
44). The corresponding solvation parameters γ and b are 0.00542 Kcal/molÅ
2 and 0.92 Kcal/mol, respectively for the PB method, and 0.0072 Kcal/molÅ
2 and 0 Kcal/mol respectively, for the GB method. The probe radius of the solvent was set to 1.4 Å. PARSE radii were used in solvation model (
45). The obtained conformational entropy change upon ligand binding (T∆S) from the sum of the translational, rotational and vibrational components, was computed using quasi-harmonic entropy approximation. In this study, the single trajectory approach was applied to estimate the energies. This means that the protein and ligand geometries were taken from protein-ligand complex, thus there is no internal energy contribution to the net molecular-mechanical binding energy (∆E
gas). Estimation of energies in this manner has been proven successful in many studies (
46). The separate trajectory approaches in which three trajectories of complex, free receptor and free ligand used for energy calculations were deficient in practice due to the limitations of sampling and large fluctuations.