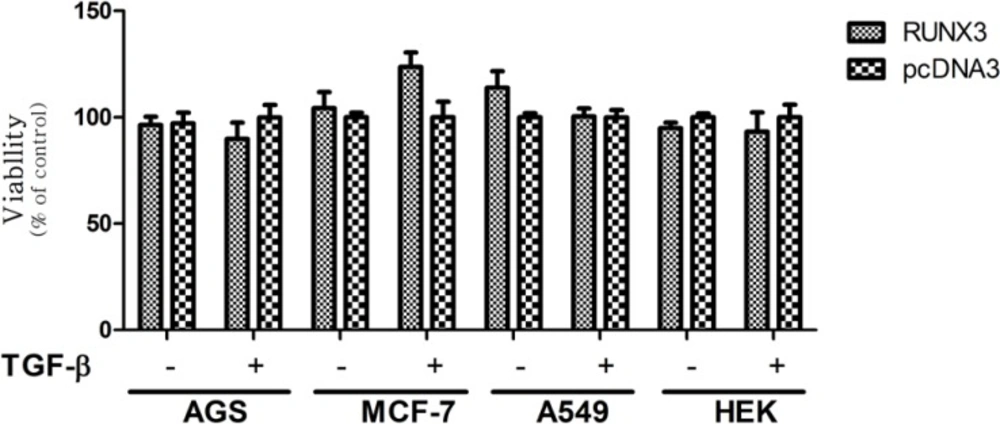
Previous studies have pointed to the growth inhibitory effect of RUNX3 in different cell lines, including AGS, MCF-7, A549, and HEK 293(
36-
38). However, with regard to our results, the RUNX3 did not notably induce growth inhibition in these four cell lines in the presence or absence of TGF-β (
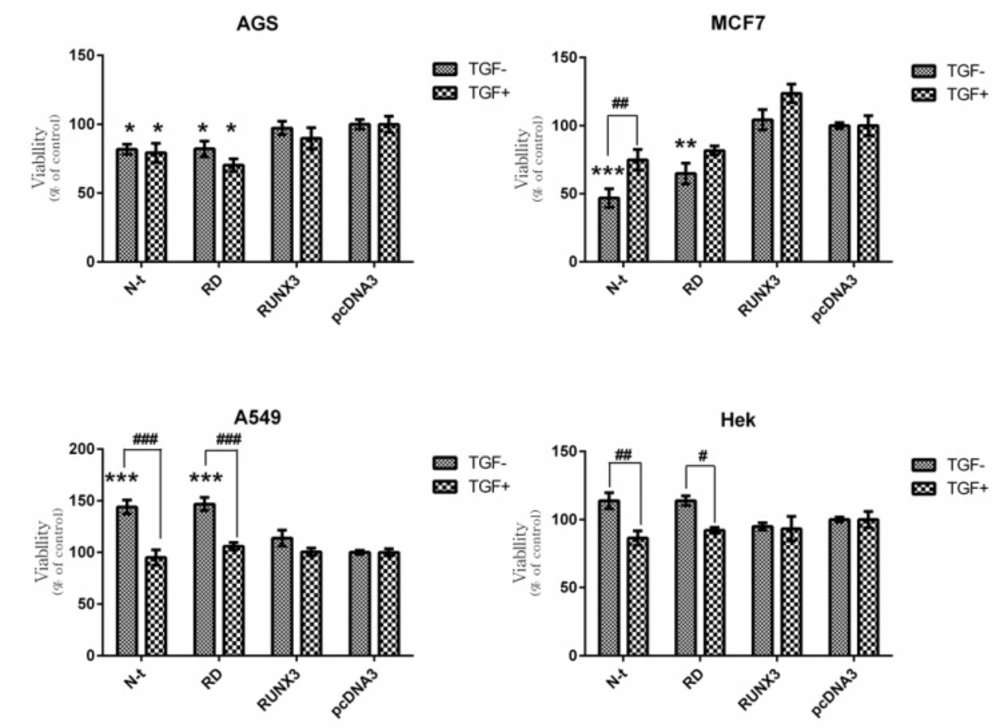
Figure 3). Furthermore, the N-t and RD truncates showed different trends in these cell lines. Cell proliferation in the AGS and MCF-7 significantly decrease while in the A549 significantly increase. On the other hand, transfection of N-t and RD did not considerably affect the cell proliferation in the HEK293.
Given the above mentioned results stated thus far, a number of questions are rising:
Why RUNX3, which is known as an important tumor suppressor, does not exert any notable growth inhibitory effect in our cell lines?
Why N-t and RD constructs have a significant effect on cell proliferation, while the full-RUNX3 does not exert a such effect?
Why N-t and RD constructs provoke divergent effects on each cell-line?
Why each cell-line shows different behaviour in the presence of TGF-β?
For the first time, Ito team established that Runx3 was expressed in the glandular stomach epithelial cells, and Runx3 null gastric mucosa developed hyperplasia owing to the promotion of proliferation and suppression of apoptosis in epithelial cells. Consequently, they proposed that RUNX3 is a tumor-suppressor gene causally involved in gastric carcinogenesis (8). After publishing these data, many other papers substantiated the onco-suppressory role for RUNX3 in gastric and other tissues. By contrast, Groner team, which are pioneer in RUNX researches ruled out Itoʼresults. They generated RUNX3
-/
- mice, which did not show any early-onset gastric hyperplasia and did not develop gastric carcinoma (
14,
39,
40). They also assessed RUNX3 expression by various techniques, including IHC with eight different anti-RUNX3 antibodies and showed that RUNX3 was not even transiently expressed at any time point in the gastrointestinal (GI) tract epithelium (
14). Furthermore, in their previous study of RUNX expression pattern during embryogenesis, they demonstrated that Runx3 expression was confined to mesenchymal elements, whereas Runx1 was expressed in both epithelium and mesenchyme cells (
41). They concluded that absence of RUNX3 expression in normal epithelium undermines the tumor suppressor role for RUNX3. This paradigm was supported by Cravalho
et al., who likewise reported lack of RUNX3 expression in human normal GI tract epithelium (
16). Moreover, Friedrich
et al. examined RUNX3 expression in gastric biopsies from 105 patients. The IHC results indicated RUNX3 protein expression only in infiltrating leukocytes, but not in the gastric epithelium (
15). Additionally, Itoʼs lab could not repeat their results on RUNX3-lacZ staining in GI tract (
42). All the cell lines we used in this study are epithelial cells. Considering Gronerʼ results, RUNX3 is not expressed frequently in epithelial cells and hence, we cannot anticipate a tumor suppressor role for RUNX3 in these cell lines. On the other hand, RUNX is not merely a strong transcription factor, rather it can recruit other cofactors to exert its function as a scaffold (
5). This notion can partly justify why we did not observe any significant effect of RUNX3 on cell proliferation. If so, one can ask why the N-t and RD constructs of RUNX3 have significant effects on cell proliferation, while the full-RUNX3 does not induce such effect.
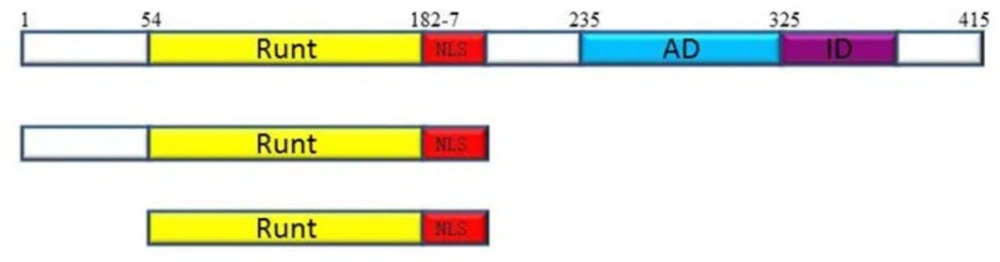
The Runt domain is a conserved DNA binding domain (128aa) with more than 90% identity among three RUNX genes. This domain is considered as the main part of RUNX proteins since, only this part binds to a specific motif in DNA (
32). Furthermore, Runt domain also contributes to nuclear localization and is able to translocate to the nucleus and bind to DNA with stronger affinity compared to the full protein (
29). Although the activation domain is responsible for modulating RUNX function through interaction with certain cofactors, the exposed Runt domain by itself can interact with many cofactors, including Ets, C/EBP, and cbfb which consequently can regulate a number of RUNX functions regarding the cell context (
5,
43,
44). Accordingly, the N-terminal constructs of the RUNX can interfere with the activity of all three mammalian RUNX proteins due to its high identity among three RUNX genes as well as its higher affinity for the consensus DNA binding site. Considering these data, the N-t and RD might bind to all three RUNX proteins’ DNA binding site and interfere with or mimic the activity of RUNX1 and RUNX2 in addition to the RUNX3.
RUNX transcription factor participates in some biological activities of TGF-β signalling (
45). TGF-β signalling pathway has a major role in the regulation of cell proliferation and differentiation which includes both tumor-suppressor and proto-oncogene arms. Even in the same cells, the response of TGF-β pathway differs depending on the environmental factors and cellular context. Hence, TGF-β may act as tumor suppressing or tumor promoting factor in cancer development. It has been generally established that TGF-β is a tumor-suppressor in the early stages of carcinogenesis, whereas is a tumor-promoter at late stages in tumor progression (
46-
48). RUNX proteins take part in both arms of the TGF–β pathway (
45,
49). In fact, RUNX proteins show context-dependent manner in cell proliferation and differentiation; thus, are either proto-oncogenes or tumor-suppressors regarding their context (
50).
Given the notion that the AGS cell-line is resistant to TGF-β, as anticipated, the presence or absence of TGF-β did not significantly alter cell proliferation in AGS transfected cells. Moreover, in this cell-line, the N-t and RD could significantly decrease cell proliferation (Figure 4a). It is known that RUNX1 is expressed in gastric epithelial cells and functions as a tumor-suppressor in gastric cancer cells (
51). It is possible that these two truncated forms mimic the RUNX1 function in this context. Since AGS is resistant to TGF-β, this effect is likely due to other pathways that RUNX takes part in, such as FOXO and Wnt signalling (
52,
53). Similar to gastric epithelium, RUNX1 is expressed in breast epithelial cells and acts as a tumor suppressor in breast cancer(
3,
54). In MCF-7 cell-line, the N-t and RD constructs considerably decreased cell proliferation in the absence of TGF-β, while the interaction of TGF-β with these constructs reduced their inhibitory effect (
Figure.4b). This means that in the presence of TGF-β, these truncated forms simultaneously show negative and positive levels of regulation in cell proliferation through different signalling pathways. In ERα-positive breast cancer cells like MCF-7, the tumor suppressor arm of TGF-β is impaired while, the proto-oncogene arm is functional; thus TGF-β promotes tumor development (
34). RUNX1 in TGF-β signalling pathway can participate in both tumor-suppressor and tumor-promoter arms. Additionally, RUNX1 can exert the tumor suppressor role in breast cancer through other pathways, such as FOXO (
34,
55). Consequently, the N-t and RD truncates, like RUNX1 can perhaps take part in pathways, such as FOXO in the absence of TGF-β, while in the presence of TGF-β, beside FOXO pathway, can play a weaker role in tumor-promoter arm of TGF-β.
Interestingly, the N-t and RD constructs could notably provoke cell proliferation in A549 cells (
Figure 4c). Consistent with this finding, it has been reported that RUNX2 plays pivotal role in tumorogenicity in non-small cell lung cancer (NSCLC) (
56,
57). Hence it is tempting to speculate that the N-t and RD perhaps mimic the RUNX2 function in NSCLC A549 cell line. On the other hand, the effect of these two constructs was completely blocked by TGF-β. In line of our result, Kang
et al.(
58) have demonstrated that TGF-β can block RUNX2 function, which can justify our results.HEK is a non-tumorogenic embryonic cell-line possessing the intact signalling network for RUNX and TGF-β pathways. Consequently, transfection of N-t and RD did not considerably affect the cell proliferation either in the presence or absence of TGF-β (
Figure 4d).