Plant material and extracts
In this study, the leaves of Ilex spinigera were collected from Mazandaran forests in September 2012 and authenticated by Dr. Alireza Naghinezhad. The voucher specimen was deposited at the Herbarium of the Department of Biology, University of Mazandaran (voucher Nr.4034). Extraction was carried out by maceration method. The leaves were cleaned and dried at room temperature in Laboratory and then in an oven at 60 °C for 24 h after which the dried plant material was ground to powder using an electric mill to obtain uniform size particles. Two gs of plant sample were extracted with 3 × 50 mL of methanol, ethanol and distilled water as solvent on shaker at room temperature. Each extract was centrifuged and filtered through Whatman No. 1 filter paper. The filtrate was evaporated to dryness in vacuo at 40 °C in a rotavapor. The dried sample of each extract was weighed to determine the yield of soluble constituents and stored at 4 °C until use.
Chemicals and reagents
1, 1-diphenyl-2-picrylhydrazyl radical (DPPH˙), ascorbic acid (AA), quercetin, gallic acid, folin-ciocalteu reagent, trichloroacetic acid (TCA), butylated hydroxyl toluene (BHT), ammonium molybdate, aluminium chloride, potassium ferricyanide (K3[Fe(CN)6]), and TMB (3, 3′, 5, 5′-Tetramethylbenzidine), HRP-enzyme, chloramine T trihydrate, hydrogen peroxide (30%), Tris-Cl buffer, Triton X100, Sodium dodecyl sulfate (SDS), MgCl2, AAPH [2, 2'-azobis (2-amidinopropane hydrochloride)], TBA (Thiobarbituric acid), n-butanol, PBS [Na2HPO4 (8.1 mM), NaH2PO4 (1.9 mM), EDTA (10.0 μM)] and all chemicals were of analytical grade and purchased either by Fluka, Sigma or Merck Co.
Determination of total phenol and flavonoid contents
Polyphenols (compounds that generally characterized with the presence of several hydroxyl groups attached to a ring or multi-ring structures) are classified as phenolic acids, stilbenes, lignans and flavonoids are the major plant compounds with antioxidant activity (
26). Therefore, it is important to measure their amount in natural extracts. The amount of total phenolic compounds was determined according to Folin-Ciocalteu method (
27). This assay was carried out following the same method as reported previously. A calibration curve of gallic acid was prepared, and the result for the extracts was expressed as µg GAE (Gallic acid equivalents/100 g extract) (
28). Aluminum chloride colorimetric method was used for determination of flavonoid contents of
I.spinigera extracts. Total flavonoid contents were calculated as quercetin from a calibration curve. The calibration curve was prepared by preparing quercetin solutions as concentrations 5-50 µg/mL (
29).
Antioxidant activity determination by DPPH radical scavenging assay
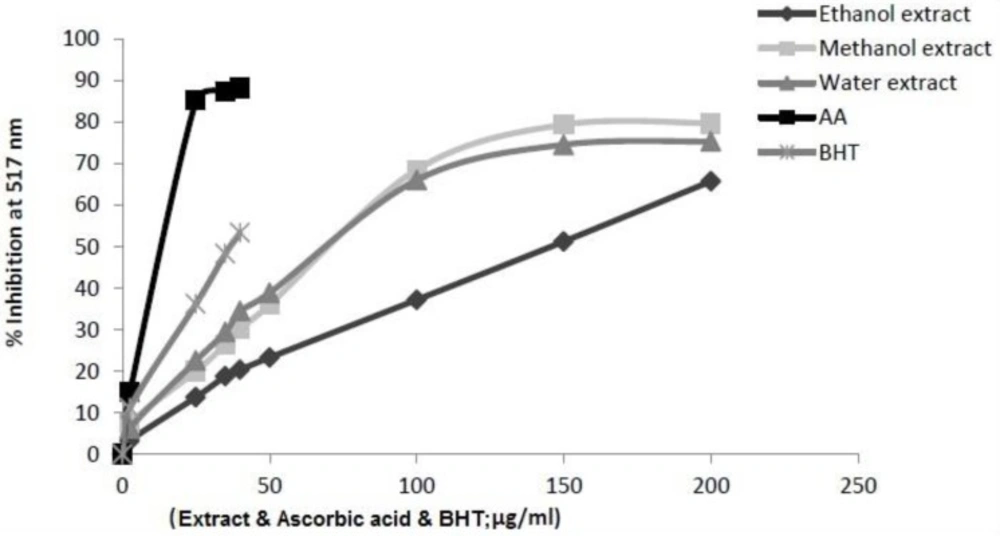
The ability of the extracts to scavenge DPPH radicals was determined according to the method of Ardestani (
30). 1 mL of a 1 mM methanolic solution of DPPH˙ was mixed with 3 mL of each extract solution in methanol (containing 25-200 µg/µl of dried extract). The mixture was then vortexed vigorously and left for 30 min at room temperature in the dark. The absorbance was measured at 517 nm and anti-DPPH activity was expressed as percentage DPPH scavenging relative to control using the following equation:
Where Acontrol was the absorbance of the blank tube and Asample was the absorbance of I.s. or standard. Ascorbic acid and BHT were used as positive controls. The DPPH solution without sample solution was used as a control. Triplicate samples were run for each set and averaged. The results were given as mean values ± SD.
Total antioxidant capacity
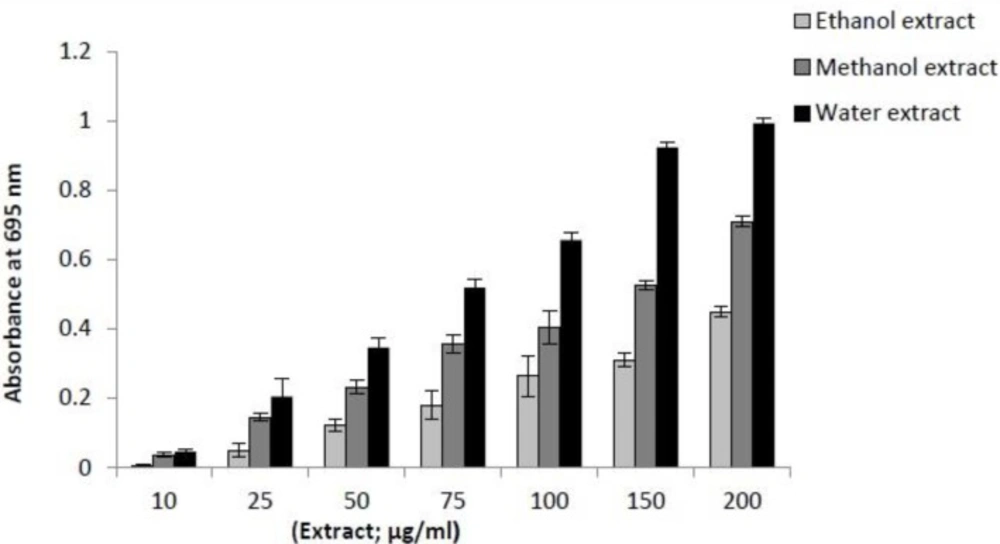
The antioxidant activity of the extracts was evaluated by the phosphomolybdenum method according to the procedure of Prieto (
31). This assay is based on the reduction of Mo (VI) to Mo (V) by the sample and the subsequent formation of a green phosphate/Mo (V) complex at acidic pH. In this test different concentrations of each extracts (10-400 μg/μL) were used. An aliquot of 0.1 mL of sample solution was combined with 1mL of reagent solution (0.6 M sulphuric acid, 28 mM sodium phosphate, and 4 mM ammonium molybdate). The tubes were capped and incubated in a water bath at 95 °C for 90 min. After the samples were cooled to room temperature, the absorbance of each test tube was measured at 695 nm.
Antioxidant activity determination by Reducing Power
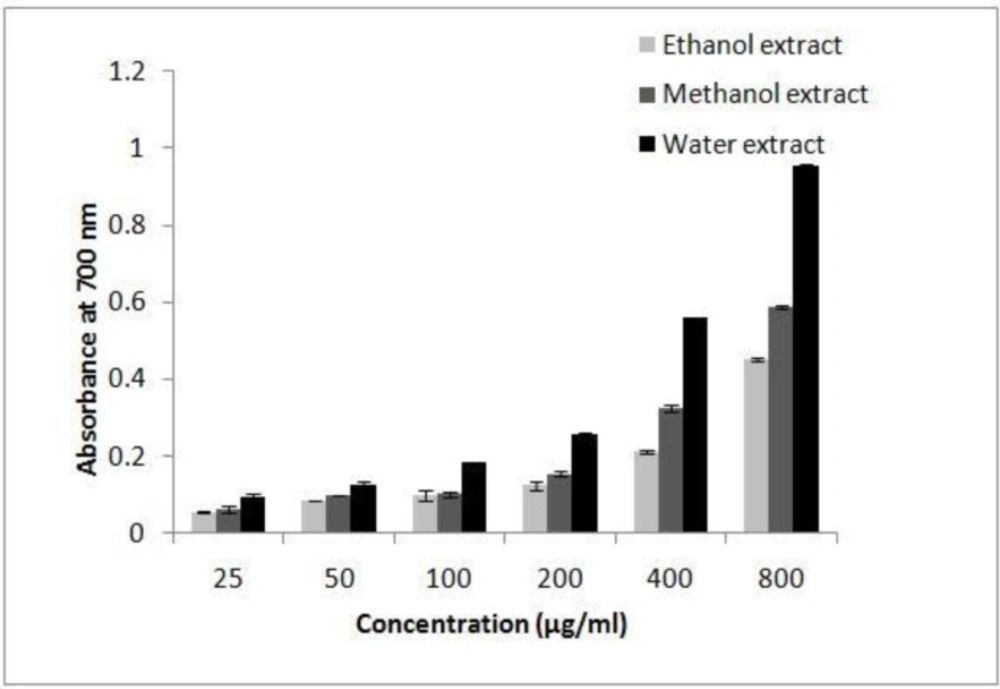
The reducing power of extracts was determined according to the method of Yildirim (
32). Different amounts of each extracts (25-800 μg/μL) in 1 mL of distilled water were mixed with phosphate buffer (2.5 mL, 0.2 M, pH 6.6) and potassium ferricyanide [K
3Fe(CN)
6] (2.5 mL, 1%). The mixture was incubated at 50
oC for 20 min. A portion (2.5 mL) of trichloroacetic acid (10%) was added to the mixture to stop the reaction. The upper layer of the solutions (2.5 mL) was mixed with 2.5 mL of distilled water and FeCl
3 (0.5 mL, 0.1%), and the absorbance was measured at 700 nm. Increased absorbance of the reaction mixture indicated increased reducing power of the extracts. Ascorbic acid was used as positive control.
Protective effect on DNA damage-induced by AAPH
DNA extraction from Cow Blood
DNA was extracted from cow blood using general phenol-chloroform extraction method (
33,
34). The extracted total DNA was measured by the following equation: [dsDNA] = OD
260 × 50 × 1/Dilution and estimated its molecular weight by electrophoresis on an agarose gel (1%) at Voltage 90 V for 2 h. The concentration of DNA was expressed as the millig of DNA per milliliter of PBS. The DNA bands were stained with 1μg/mL ethidium bromide and visualized by UV-transilluminator. Banding was photographed using a Gel-doc system.
The antioxidant effect of I.s. extracts on AAPH-Induced oxidation of DNA
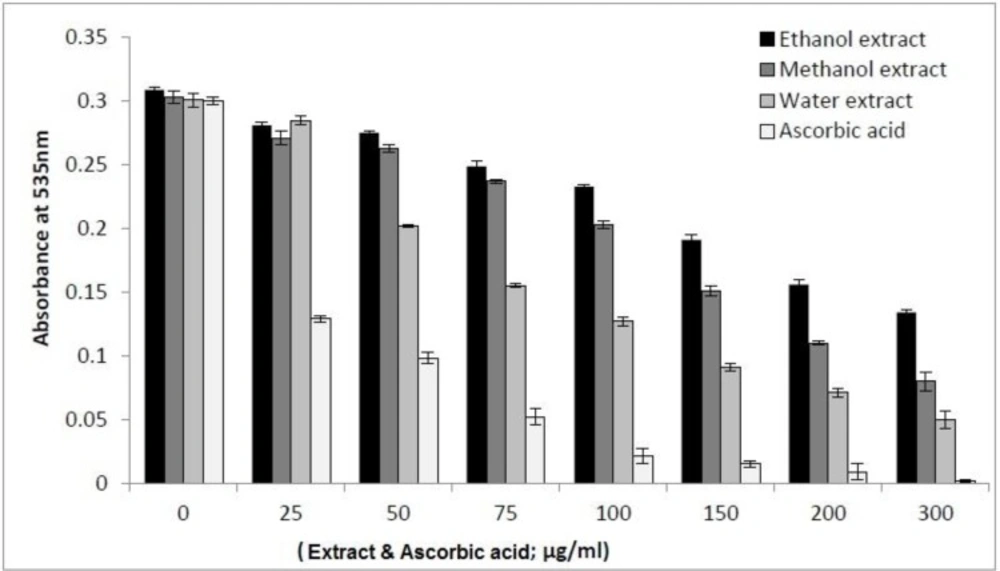
AAPH-induced oxidation of DNA was performed according to the Zhao
et al. method with a slight modification (
17,
35). Briefly, various concentrations of plant extracts in DMSO were added to PBS solutions of AAPH and DNA, in which the final concentration of DNA and AAPH was kept 2.0 mg/mL and 40 mM, respectively. Then, the extracts were dispatched into test tubes with a final volume of 2.0 mL. Then the tubes were incubated in a water bath at 37 ˚C to initiate the oxidation. Three tubes were taken out at appropriate interval and cooled immediately, to which 1.0 mL of TBA (1.00 g TBA and 0.40 g NaOH dissolved in 100 mL PBS) and 1.0 mL of trichloroacetic acid aqueous solution 3% was added. The tubes were heated in a boiling water bath for 15 min. After cooling, 1.5 mL of n-butanol was added and shaken vigorously to extract TBARS. The absorbance of n-butanol layer was measured by a spectrophotometer at 535 nm. Different concentrations of ascorbic acid were used as positive controls.
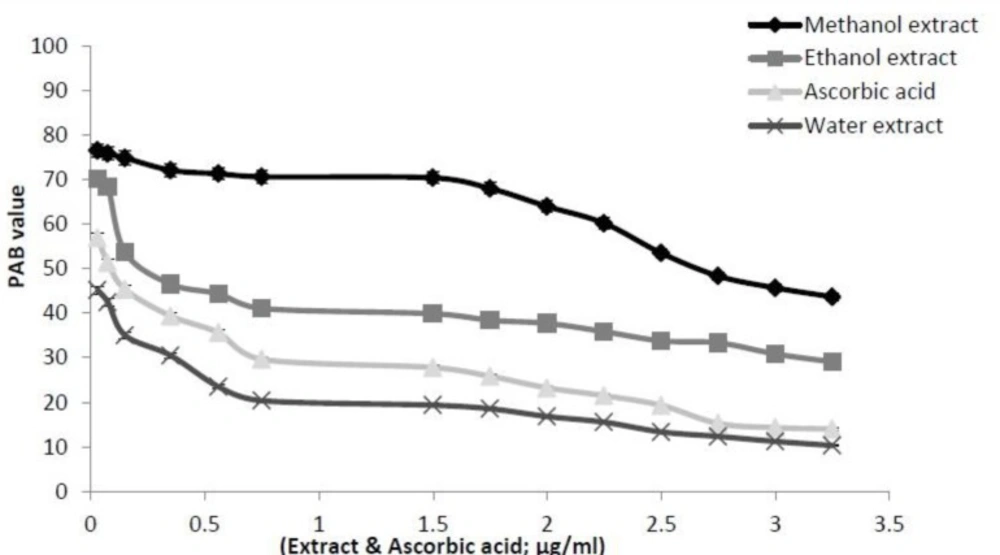
Prooxidants–antioxidants balance (PAB) assay
A modified PAB assay was applied according to the method of Alamdari (
4). Sixty mg TMB powder was dissolved in 10 mL DMSO; for preparation of TMB cation, 400 μL of TMB / DMSO was added in 20 mL of acetate buffer( 0.05 M; pH 4.5), and then 70 μL of fresh chloramine T (100 mM) solution was added into this 20 mL, mixed well, incubated for 2 h at room temperature in a dark place; 25U of peroxidase enzyme solution was added into 20 mL TMB cation, dispensed in 1 mL and put at -20 °C; in order to prepare the TMB solution 200 μL of TMB / DMSO was added into 10 mL of acetate buffer [0.05 M buffer, pH 5.8]; the working solution was prepared by mixing 1 mL TMB cation with 10 mL of TMB solution, incubated for 2 min at room temperature in a dark place and immediately used. 10 μL of each sample with various concentration (0.03-3.25 μg/μL), standard or blank (distilled water) were mixed with 200 μL of working solution in uncoated micro titer plate, which was then incubated in a dark place at 37 °C for 12 min. The reaction was stopped by the addition of the stop solution (100 μL of 2N HCl) was added to each well the absorbance and measured in an ELISA reader (Biotek ELX800) at 450 nm. A standard curve was provided from the values relative to the standard solution. The values of the PAB are expressed as percentage of hydrogen peroxide in the standard solution. The standard solutions were prepared by mixing varying proportions (0-100%) of 250 μM hydrogen peroxide with 3 mM uric acid (in 10 mM NaOH). The values of unknown samples were then calculated based on the values obtained from the above standard curve.
Statistical analysis
In-vitro experimental results were given as mean ± standard deviation of three parallel measurements. The experimental values were evaluated by using one-way analyses of variance. P values < 0.05 were regarded as significant.