COX-2 signaling pathway has been addressed as an indispensable mediator in the pathogenesis of a variety of malignancies, in particular lung cancer (
6,
7). Consistent with this notion, it has been appeared that COX-2 pathway blockade can be a desirable approach in the treatment of a range of cancer models, including the lung neoplasia (
27). Previous experiments have delineated functional crosstalk between COX-2 and a subset of molecular targets, such as p53 (-). Considering these statements, the role of COX-2 inhibitors as a possible candidate to induce cell death through apoptotic machinery has been investigated in numerous reports thus far (
23,
28).
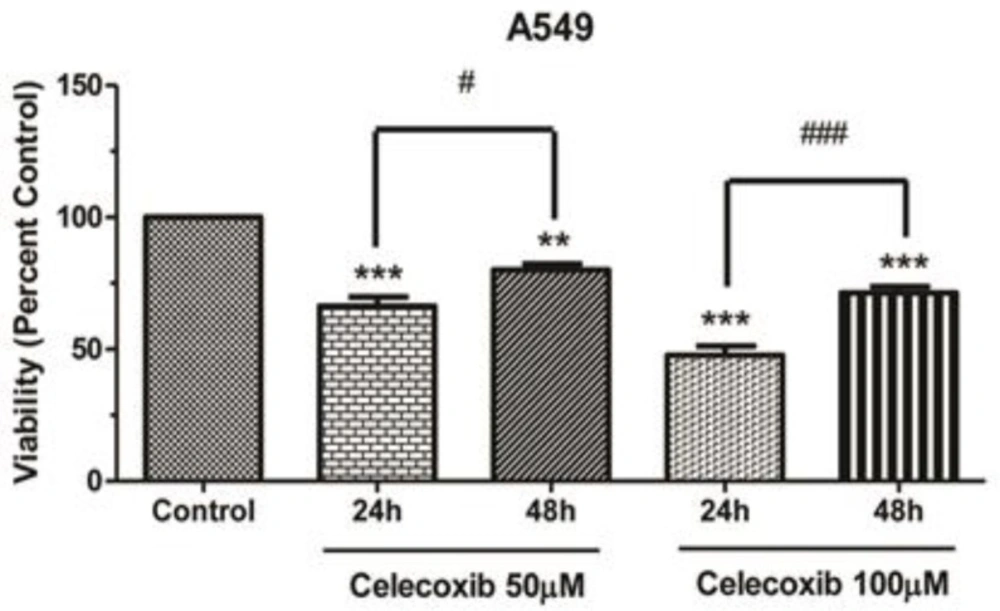
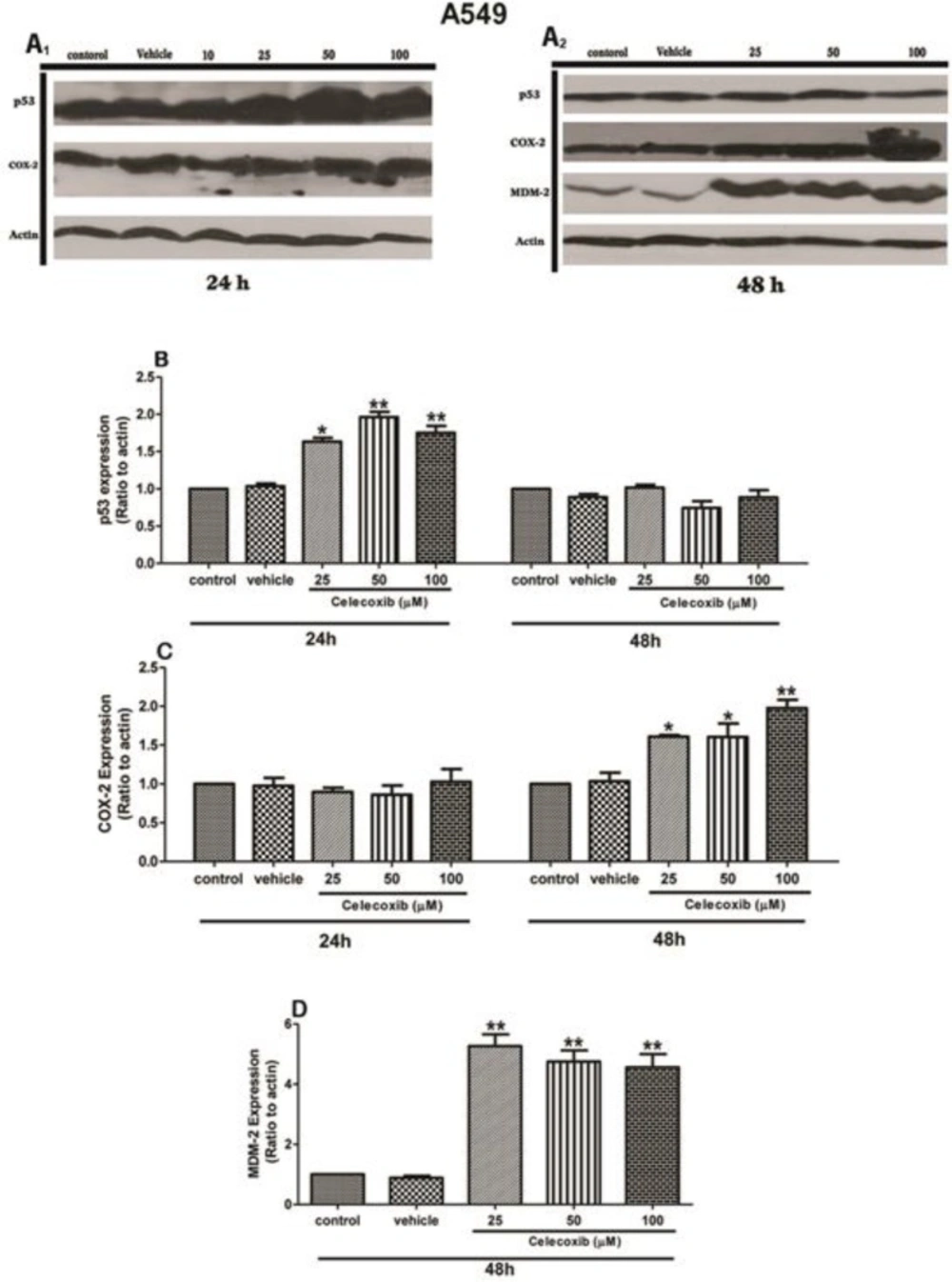
In this respect, we aimed to examine the effect of Celecoxib on p53 expression and persued p53 signaling interaction with COX-2 and MDM2 pathways upon Celecoxib treatment in A549 cells. The cell viability assay has shown that Celecoxib caused a dose-dependent growth inhibition in wild-type A549 cells within 24 h. However, prolonged exposure to the drug conversely led to increase cell viability percent, suggesting that Celecoxib treatment biphasically modulates cell growth depending on the dose and timing of the treatment. In accordance with this, our western blot results have also demonstrated that Celecoxib enhanced p53 expression within 24h in parental cells in a dose-dependent manner (1.5-2-fold of control, P values <0.05, 0.01, respectively). It has been appeared that Akt is a self-evident repressor of the p53 expression through MDM2 and consistently, several studies have reported that Celecoxib can inhibit Akt signaling pathway hence, the resultant p53 up-regulation might be ascribed to disruption of Akt signaling induced by Celecoxib(-,
29). Although Celecoxib could increase p53 level within 24h, COX-2 expression was barely affected at this time point, indicating that Celecoxib augmented p53 expression, independently of COX-2 inhibition (
Figures 2B, C). This result suggests p53 as a COX-2-independent molecular target. Considering this finding, we next sought to determine whether inhibition of COX-2 pathway affects prolonged p53 signaling. Paradoxically, Celecoxib exposure lowered p53 expression within 48h, so that p53 returned to the control level at this time point compared to the corresponding 24 h (
Figure 2B). Coincidentally, Celecoxib strikingly could result in COX-2 induction at 48 h (
Figure 2C).The resultant COX-2 elevation upon Celecoxib treatment can be attributed to p53 that can induce COX-2 expression through Ras/Raf/MAPK cascade according to the prior experiments(
24). Accompanying this, MDM2 expression was markedly augmented at 48h, indicating a direct correlation between these signaling pathways (
Figure 2D). Thus, it seems that p53 triggers a negative regulatory feedback which finely tunes the cellular homeostasis by manipulating the expression of both MDM2 and COX-2.From the clinical standpoint, such findings can point toward the possibility that Celecoxib treatment may not be a proper therapeutic strategy in lung cancer cells owing to its potential role in the activation of counter-regulatory pathways, resulting in the induction of crucial oncogenes, such as COX-2 and MDM2. In supporting this issue, a recent investigation has also substantiated that Celecoxib promotes cell invasion and confers cells resistant to chemotherapy through activation of MEK-ERK signaling cascade (
30). Consistently, it has been appeared that ERK promotes tumorogenesis via MDM2 signaling axis (
31).Conceivably, blockage of MDM2 and/or other contributing mediators induced by Celecoxib might be an effective approach to hamper Celecoxib-induced cancer promotion, although elucidating such assumptions demands further investigations. Taken together, such a scenario underscores intensive preclinical assessment before applying COX-2 inhibitors in the treatment of lung tumors.