Bacterial growth and extract treatment
For this study,
S. mutans ATCC 35668 was purchased from the Industrial Fungi and Bacteria Collection Center (Tehran, Iran). Bacterial suspension was prepared using a pure culture of this bacterial strain, grown in brain heart infusion (BHI) broth (Merck, Germany) medium. The inoculated medium was incubated at 37˚C. After this time, broth culture was inoculated in 8.5 % saline solution to obtain an optical turbidity of 1 McFarland standard. In this study, the water extracts of
Punica granatum L. flowers and
Rhus coriaria L. fruit, prepared in our previous studies, were used (
16,
17).The stock of extracts was prepared with a concentration of 4.68 mg/mL and dissolved in deionized water. Prepared solutions were filter sterilized through a 0.2 μm pore-size polycarbonate filter. Then, extract solutions in a final MBIC (0.39 mg/mL for
Rhus coriaria L. and 6.125 mg/mL for
Punica granatum L.) were added to BHI broth medium in sterile test tubes. These tubes were then inoculated with the bacterial suspension prepared equivalent to 1 McFarland. The tubes were incubated overnight.
RNA extraction and cDNA synthesis
Total RNA was extracted from planktonic cultures of non-treated (control) and 24 h treated
S. mutans with MBIC concentrations of
Rhus coriaria L. (0.39 mg/mL) and
Punica granatum L. (6.125 mg/mL) (
Table 1) (
16,
17). Before extraction, bacterial cultures (three cultures for each group; n = 3) were mixed with 2 volumes of RNA protect solution (Qiagen, USA) in order to stabilize bacterial RNA. After 5 min of incubation at room temperature, bacterial cells were collected by centrifugation (5000 g, 10 min). The cells were disrupted with re-suspension of the pellets in TE buffer (10 mM Tris·Cl, 1 mM EDTA, pH 8.0) containing 1 mg/mL lysozyme (Sigma-Aldrich, Germany) and incubation at 37˚C for 20 min. Then, the bacterial total RNA was isolated and purified using High Pure RNA Isolation Kit (Roche, Germany) according to the manufacturer’s instructions. The extraction procedure contained on-column digestion step with RNase-free DNase I in order to eliminate the residual contaminating DNA. The RNA concentration was determined spectrophotometrically using the NanoDrop instrument (mySPEC, Austria). The quality and integrity of the RNAs were determined by measuring OD260/280 ratio and agarose gel electrophoresis of RNA respectively.
| Test agent | MBC1(mg/mL) | MIC2(mg/mL) | MBIC3(mg/mL) |
|---|
| Punica granatum L. | 100 | 50 | 6.125 |
| Rhus coriaria L. | 6.125 | 1.56 | 0.39 |
[object Object]
[object Object]
[object Object]
In order to synthesize cDNA, 1 µg of isolated RNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA) according to the manufacturer’s instructions in the presence of random primers on a PCR amplification machine (PeQlab, USA). For additional control for residual genomic DNA contamination, the cDNA synthesis reaction was carried out with each purified total RNA sample in the absence of reverse transcriptase enzyme and served as negative control.
Real-time quantitative PCR and gene expression analysis
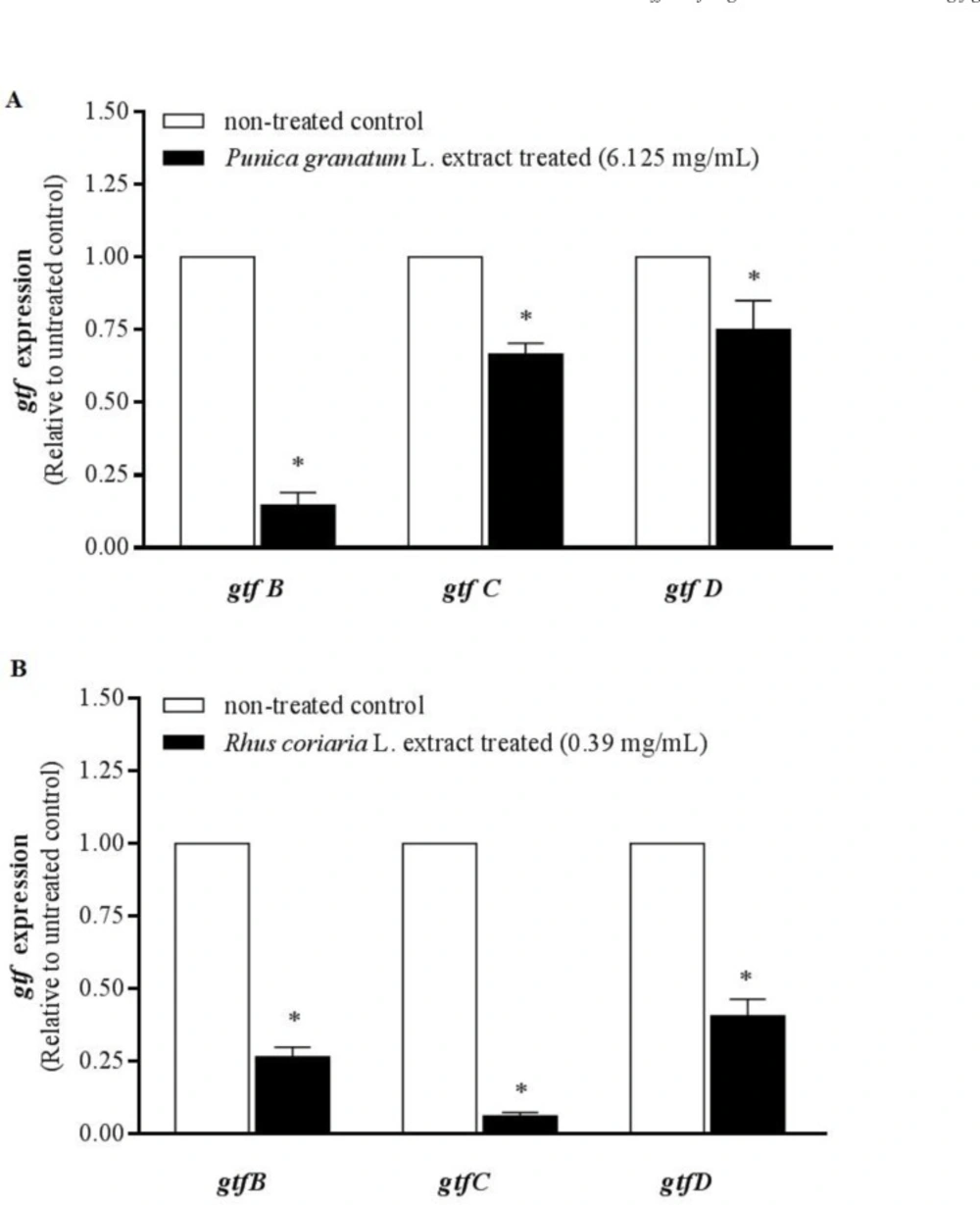
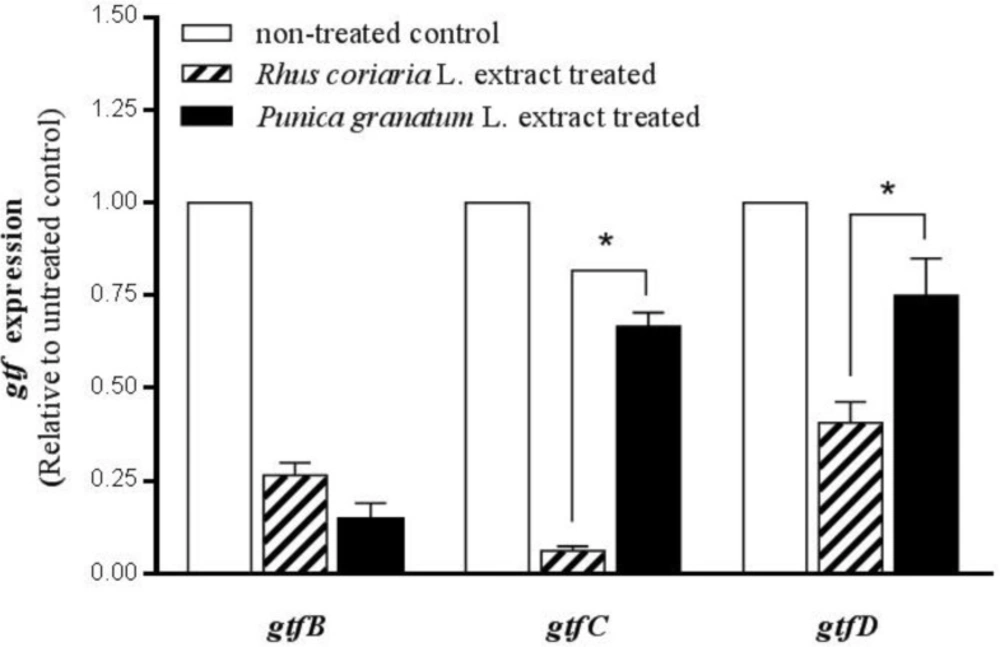
In order to quantify the changes in
gtfB,
C and
D mRNA gene expression in
S. mutans exposed to the two understudy extracts in comparison with the control (non-treated
S. mutans), real-time PCR was performed. All primers (
gtf genes and housekeeping genes) for real-time PCR were controlled with NCBI Primer Blast software and obtained commercially from Takapuzist Company (Bioneer, Korea) (
Table 2).
| Gene | Sequence (5´→3´) | Product Size (bp) |
|---|
| gtfB | Forward: AGCAATGCAGCCAATCTACAAATReverse: ACGAACTTTGCCGTTATTGTCA | 96 |
| gtfC | Forward: GGTTTAACGTCAAAATTAGCTGTATTAGCReverse: CTCAACCAACCGCCACTGTT | 91 |
| gtfD | Forward: ACAGCAGACAGCAGCCAAGAReverse: ACTGGGTTTGCTGCGTTTG | 94 |
| 16sRNA | Forward: CCTACGGGAGGCAGCAGTAGReverse: CAACAGAGCTTTACGATCCGAAA | 101 |
| recA | Forward: GCGTGCCTTGAAGTTTTATTCTTCReverse: TGTTCCCCGGTTCCTTAAATT | 75 |
The resulting cDNAs and negative controls (no-RT) were amplified using the SYBR®Green PCR Master Mix (Applied Biosystems, USA). The reaction mixture (20 µL) contained 10 µL of Master Mix, 200 ng of template cDNA (2µL) and 200 nM of forward and reverse primers placed in MicroAmp® Optical tube (Applied Biosystems, USA). Three repeats of the same reaction tubes were used for each test sample and three reaction tubes without cDNA template were used as negative controls (NTC) for each primer pair in order to check for DNA contamination.
Amplification and detection of specific products were performed on the ABI StepOne™ detection system (Applies Biosystems, USA) with the following thermal cycling conditions: initial denaturation at 95˚C for 10 min (holding stage), denaturation at 95˚C for 15seconds, followed by 40 cycles of annealing and extension at 60˚C for 1 min (cycling stage). All primer pairs were checked for primer dimer by adding a melt curve stage to the end of protocol.
Threshold cycle (Cт) values were determined, and relative expression levels were calculated by StepOne ™ software version 2.1 (Applied Biosystems, USA) according to the comparative Cт (ΔΔCт) method. The quantities of cDNA for gtfB, gtfC, and gtfD genes were normalized to that of cDNA synthesized from recA gene in the same sample. These values were then compared to those obtained from the non-treated control to determine the change in gtf gene expression level in each test sample.
Statistical analysis
All real-time PCR assays for three target genes and two target samples were performed in triplicate at the same time and reproduced at least three separate experiments. The differences between the experimental group and the non-treated control group were analyzed by GraphPad Prism (version 6.01, USA). The data from fluorescent intensities were assessed by one-way analysis of variance (ANOVA) in the Tukey-Kramer post deviation test for all pairs. A P value < 0.05 was considered statistically significant.