The pEGFP-N1 plasmid (Clontech, Palo Alto, CA, USA) was purified using a QIAGEN Plasmid Mega Kit (Qiagen GmbH, Hildden, Germany). The SRL peptides (99% purity), both CLSSRLDA and LSSRLDAC sequences, were purchased from Biomatic Company (Wilmington, USA). The PAMAM G4 dendrimer was synthesized by an in-situ branch cell method, a two-step iterative process to construct poly (amidoamine) (PAMAM) dendrimers possessing either terminal ester or amine groups. The basis of the method involves (a) alkylation with methyl acrylate, and (b) amidation with ethylenediamine (
26). Lactoferrin, filipin complex (from Streptomyces filipinesis), L-polylysin (30000-70000MW) and phenylarsine oxide were bought from Sigma-Aldrich (St. Louis, MO, USA). Bifunctional PEG (NHS-PEG-MAL, MW 1000) was obtained from Nanocsinc (Boston, USA). BODIPY fluorophore (4,4-difluoro-5,7- dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionic acid, sulfosuccinimidylester,sodium salt) was purchased from Molecular Probes (Eugene, OR, USA).
Cell line
Rat Brain capillary endothelial cells (BCECs) were kindly provided from Dr. Aghaei (Neuroscience Research Center, Shahid Beheshti University of Medical Sciences), and cultured according to ATCC guidelines. BCECs were cultured in a Rat Brain Endothelial Cell Growth Medium (Cell Applications Inc, San Diego CA, USA), supplemented with 20% fetal calf serum (FCS), epidermal growth factor (100 mg/mL), L-glutamine (2 mmol/L), heparin (20 mg/mL), insulin (40 mU/mL), penicillin (100 U/mL) and streptomycin (100mg/mL) under standard conditions in a humidified 5% CO2 incubator at 37 oC.
The C6 glioma cell line was purchased from National Cell Bank of Iran (NCBI), Pasteur Institute, Tehran, Iran. C6 glioma cells were cultured in DMEM (Sigma–Aldrich), supplemented with 10% FBS, 100 U/mL penicillin and 100 g/mL streptomycin at 37 oC in a humidified incubator containing 5% CO2. All the cells used in this study were extended between 10 to 15 passages.
Synthesis of PAMAM derivatives, and Characterization of Nanoparticles
PAMAM derivatives were synthesized according to the same methods described in the previous papers (
27,
28). In brief, PAMAM-PEG-SRL at 1:6:3 ratio synthesized as previously described. And their size and physicochemical characteristics was evaluated by NMR, Zeta sizer and UV spectroscopy (
24).
Preparation of Dendrimer/DNA Nanoparticles
PAMAM and PAMAM-PEG-SRL were freshly synthesized, and diluted in distilled water. Then, a DNA solution (100 mg DNA/mL in 50 mM Sodium sulfate solution) was added to nanoparticle solution at specific weight ratio (1:10, PAMAM/DNA w/w), and gently vortexed at 37
oC for 30 sec. Afterwards, the complex was incubated at room temperature for 30 min to allow self-assembly formation. Agarose gel electrophoresis was carried out to verify the formation and stability of the nanoparticle-DNA complex (
24).
Preparation of endocytosis inhibitors
Filipin and phenylarsine oxide were dissolved in dimethyl sulfoxide (DMSO), and then diluted in PBS (pH 7.4) to obtain 0.5 mg/mL. In addition, SRL (100 mg/mL), colchicines (2.5 mM) and lactoferrin (1 mg/mL) were prepared in water and then diluted in PBS, pH 7.4 (
27).
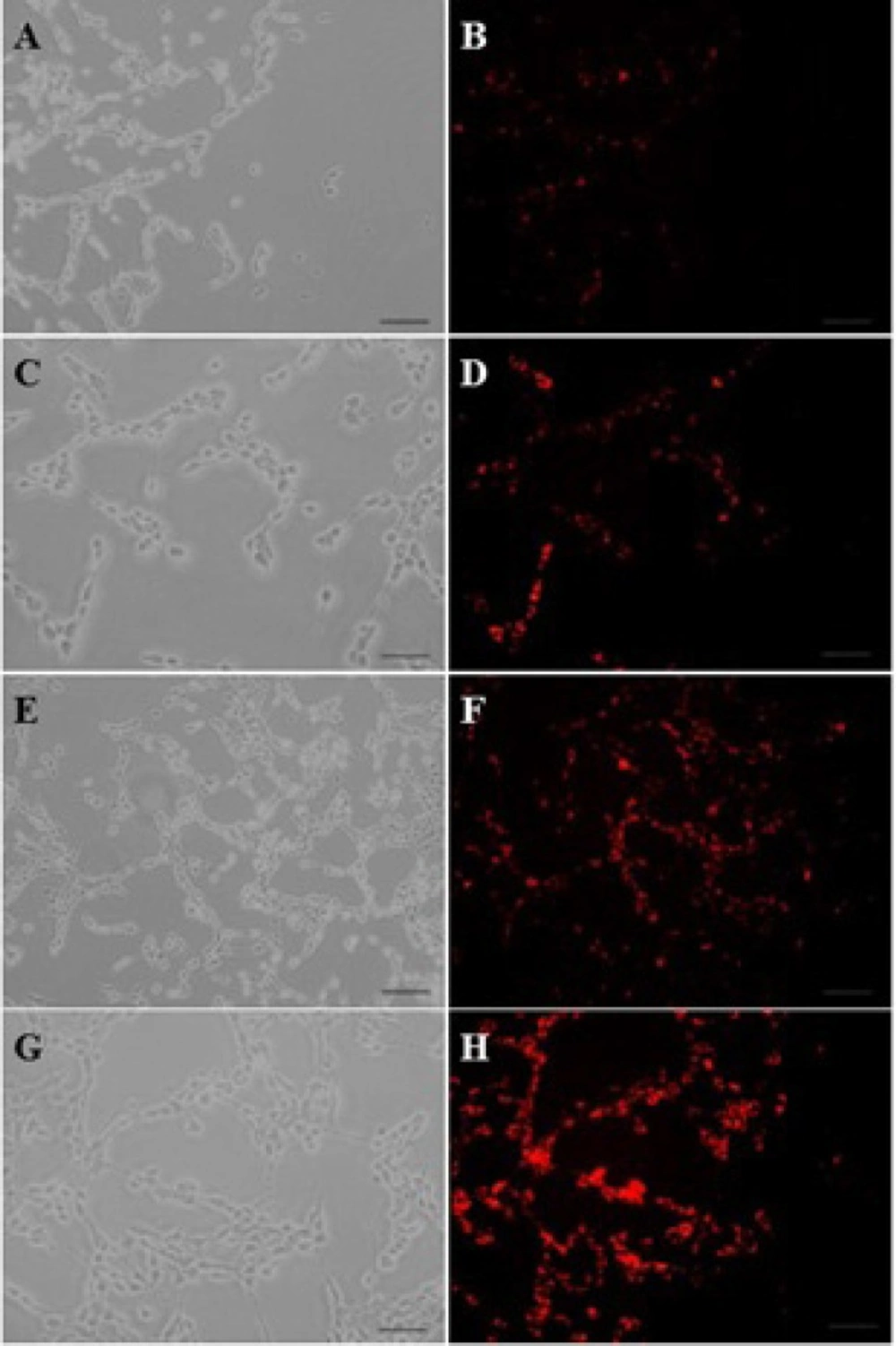
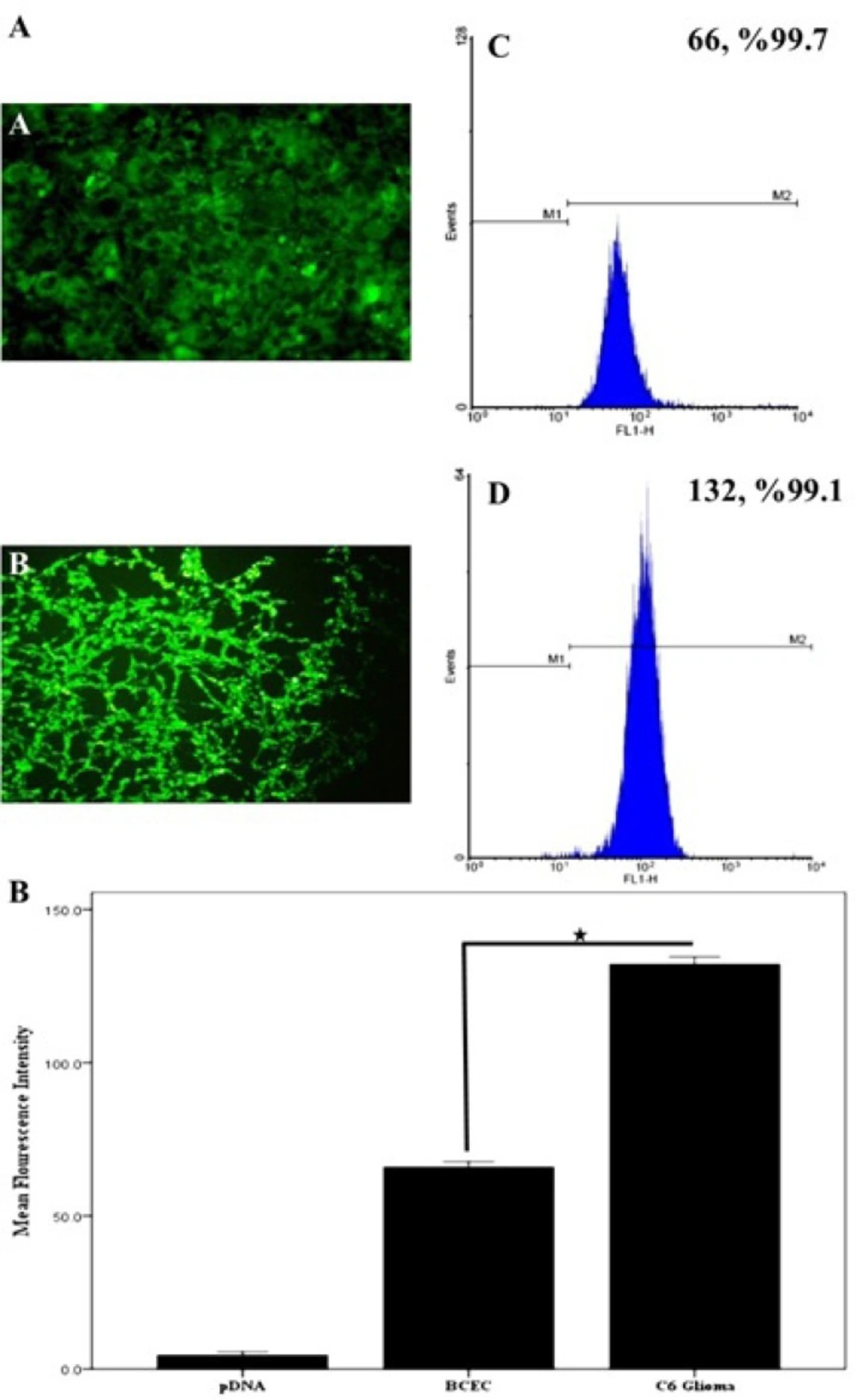
Cellular uptake of dendrimers
BCECs and C6 glioma cells were seeded at a density of 2×10
4 cells/well in 24-well plates (Corning- Coaster, Sigma, USA), for 48 hours. Cell confluence and morphology were checked under a fluorescent microscope throughout the experiment. Afterwards, the C6 glioma cells were incubated at a 1 µM concentration of BODIPY-labeled PAMAM or 0.2, 0.5 and 1.0 µM concentrations of BODIPY-labeled PAMAM–PEG–SRL at 37
oC for 30 minutes with a final concentration of 1 µM. Furthermore, to study the energy dependency of nanoparticles internalization into target cells, C6 glioma cells were exposed to BODIPY-labeled PAMAM–PEG–SRL at a density of 1µM at 4
oC. The culture supernatant was removed, and cells were then washed three times with cold PBS (pH 7.4), and examined using a fluorescent microscope (Olympus, Osaka, Japan). To quantitatively analyze the cellular uptake of PAMAM-PEG-SRL at different concentrations, the cells BCECs and C6 glioma were cultured at a density of 8×10
4 cells/well in 6-well plates (Corning-Coaster, Sigma, USA) for 72 h. After checking the cell confluence and morphology, the cells were incubated at a 1 µM concentration of BODIPY-labeled PAMAM–PEG–SRL for 30 minutes. As noted in the previous section, the cells were washed three times with PBS (pH 7.4), and then, the treated cells were trypsinized and centrifuged for 8 minutes at 1600 rpm to obtain the cellular pellet. The pellet was resuspended in PBS (pH 7.4), and analyzed by a flow cytometer (FACS Calibur, BD Biosciences, Bedford MA, USA) equipped with an Argon Ion Laser (488 nm) as a stimulating source of fluorescent. For each sample, 10000 events were collected and analyzed using WinMDI software. Cells cultured under normal conditions and live cells were considered as controls and gates, respectively. In general, the only cells at the gate were analyzed. The mean density of fluorescent cells was calculated as a histogram plot (
27).
Gene transfer efficiency into C6 glioma cells
The Plasmid DNA was covalently labeled using the fluorescent dye ethidiummonoazide bromide (EMA). Briefly, the Plasmid DNA solution (1 mg/mL in TE buffer, pH 7.0) was diluted in EMA solution until reaching a concentration of 0.1 mg/mL, and then incubated for 30 min in darkness. Then, the solution was precipitated by the addition of ethanol to a final concentration of 30% (v/v). The pellet was collected by centrifugation at 12000 rpm for 20 min, and resuspended in 50 mM sodium sulfate. EMA-DNA solution was used to prepare nanoparticles by the same method. C6 glioma cells were seeded at a density of 2.0 ×10
4 cells/well in 24-well plates, and then incubated for 48 hours. Subsequently, the nanoparticles (PAMAM or PAMAM-PEG-SRL) containing EMA-DNA were added to the cells (
27). Thirty minutes later, the culture supenatant was removed, and the remaining cells were washed 3 times with PBS (pH 7.4), and observed under a fluorescent microscope.
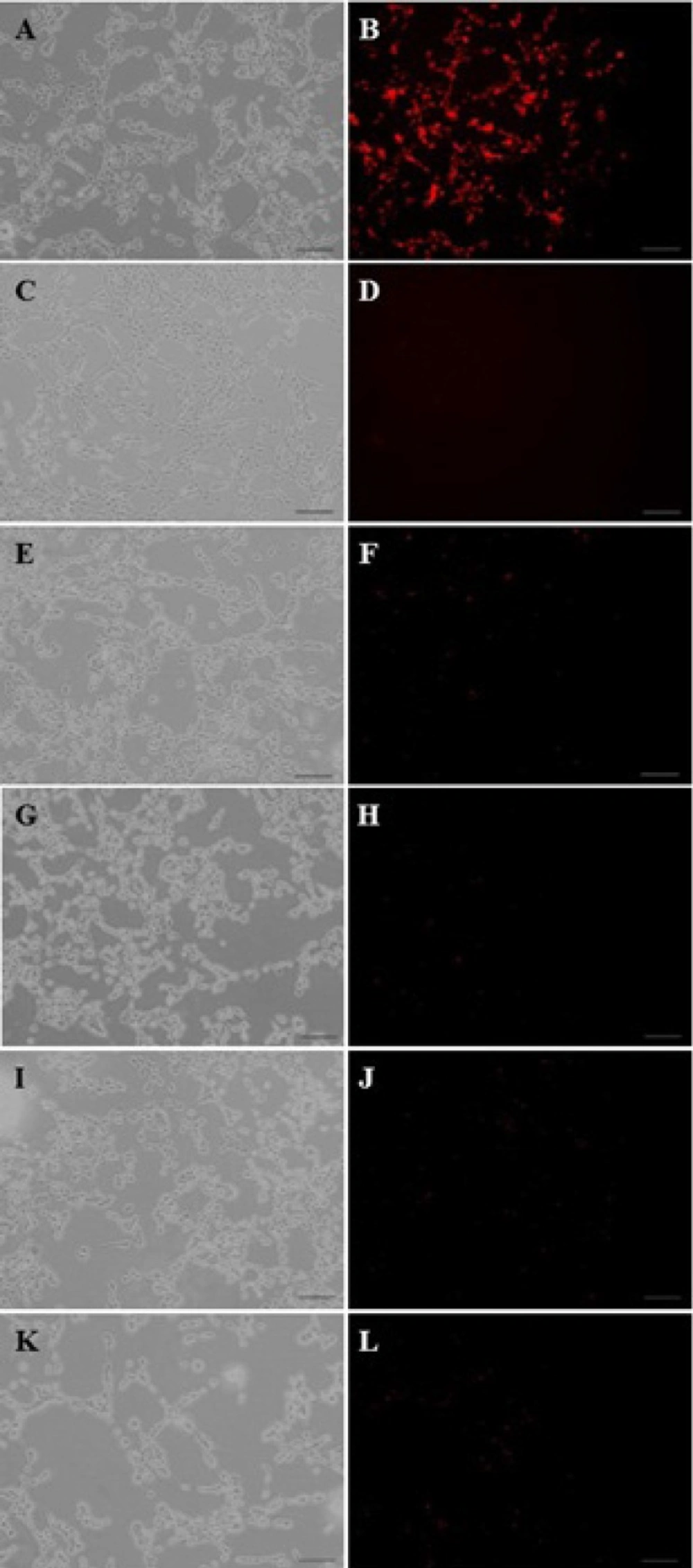
Cellular uptake mechanism of dendrimer/DNA nanoparticles
C6 glioma cells were seeded at a density of 2×10
4 cells/well in 24-well plates, and cultured using the above method. After monitoring the morphology and confluency of the complex filipin (0.5 mg/mL), phenylarsine oxide (2.5 mM), colchicines (2.5 mM) or lactoferrin (1mg/ml) were added to each well, and incubated for 10 minutes (
27). Subsequently, the supernatants were removed, and an equal volume of the nanoparticle PAMAM-PEG-SRL containing EMA-DNA were added to the above mixtures. After 30 minutes, the supernatant was removed, and the cells were washed three times with cold PBS (pH 7.4), and observed under a fluorescent microscope.
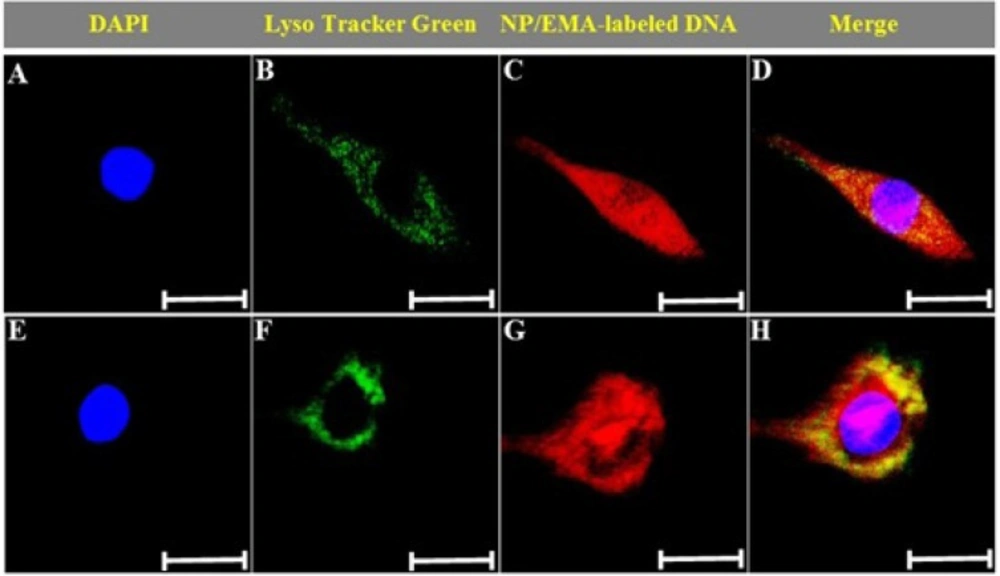
Confocal microscopy
The intracellular distribution of the nanoparticles was studied using a confocal microscopy. C6 glioma cells were seeded at a density of 10
4 cells/well in a 35 mm glass-bottom culture dish, and cultured at 37
oC under an atmosphere containing 5% CO2 for 48 hours, recultured in serum-free media, and then assessed 15 and 60 min after exposed to the PAMAM-PEG-SRL/DNA complex (5 µg DNA/well, N/P=10). After this time, the cells were washed with cold PBS, and subjected to 50nM LysoTracker Green (Molecular probes Inc.) and DAPI for 30 and 10 minutes, respectively. Afterwards, the cells were washed three times with cold PBS, fixed with paraformaldehyde and observed under confocal laser scanning microscopy (
29).
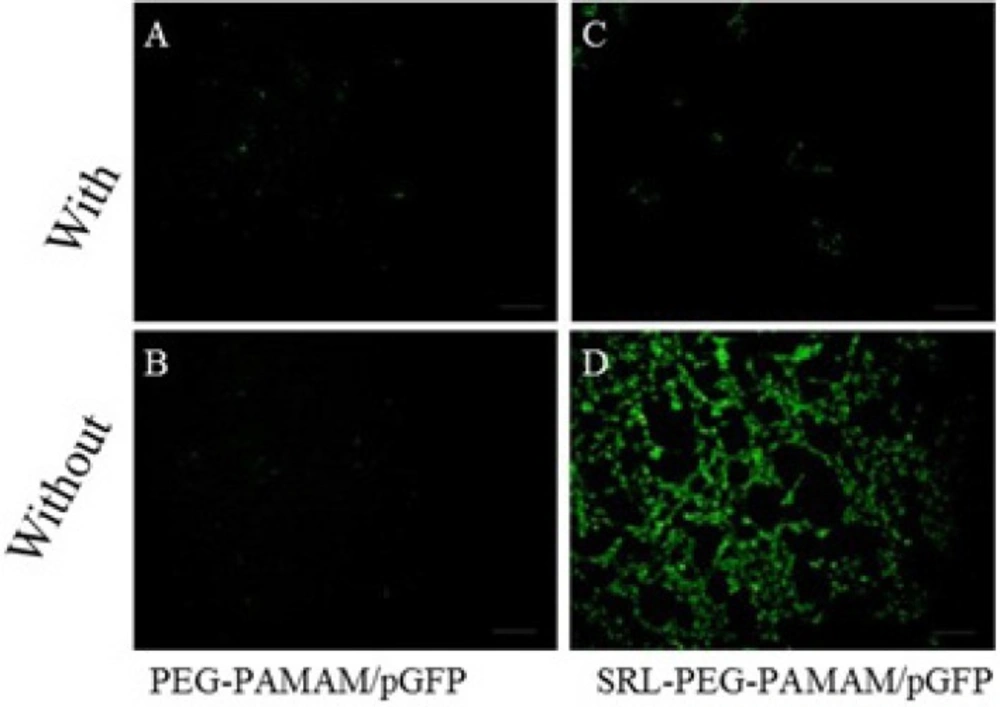
Qualitative assessment of Green Fluorescence protein expression (GFP) in transfected cells.
C6 glioma cells were cultured at a density of 1.0 × 10
6 cells/well in 6-well plates in 2 mL of culture media for 18 h to reach 50% confluence for gene therapy experiments. Prior to transfection, the medium was replaced with a serum-free medium, and the cells were then incubated for 4 h with the complexes PAMAM-PEG-SRL/DNA and PAMAM/DNA in the presence or absence of lactoferrin (8 micrograms per well, 8 μg/well) (Figure 8). The medium was replaced with a fresh medium containing 10% FBS, and then incubated again for 24 hours. GFP expression levels were detected under a fluorescent microscope (Olympus, Osaka, Japan) (
30).
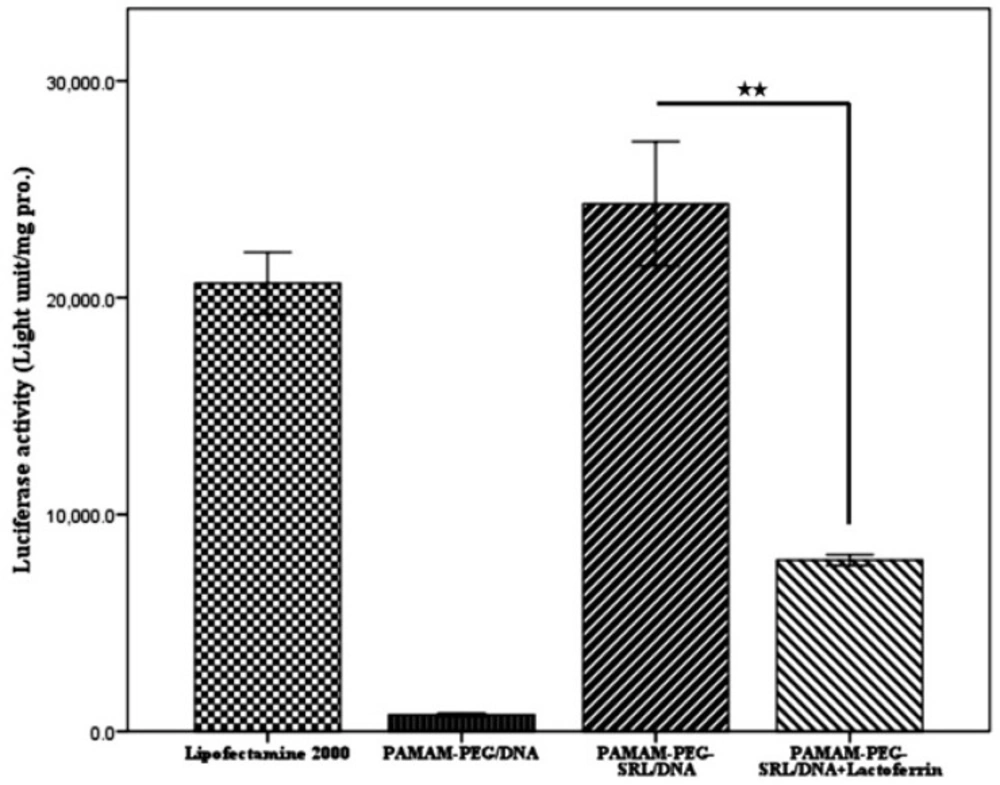
Luciferase Activity Assay.
C6 glioma cells were cultured in a 24/well plate at the appropriate confluence for 24 h, and allowed to grow 50% confluence at the transfection time. Prior to transfection, the medium was replaced with serum-free medium, and the cells were then incubated with PAMAM-PEG-SRL/pDNA complexes (4 μg /well) at a specific weight ratio under standard conditions for 4 h. Subsequently, the medium was replaced with fresh a medium containing 10% serum, and incubated again for 24 h.
To measure the luciferase activity, cells were washed with PBS two times, and 100 to 200 µL of cell lysis buffer was added to each well for 5 minutes at 37 °C. Cell lysis solution was centrifuged at 1200 rpm for 5 min, 10 µL of the supernatant was mixed with 25 µL of both luciferase substrate and ATP solution, and luciferase activity was measured with Sirius luminometer (Autolumat LB953, EG and G, Berthold, Germany). Small amounts of the protein were determined by the bicinchoninic acid assay (Bio-Rad Laboratories, Hercules, CA). Relative light units (RLUs) were normalized with the concentration of the proteins extracted from the cells. All transfection experiments were performed as triplicate. The transfection activity was reported by RLUs (
31). Lipofectamine was used as a control to compare each sample.
Statistical analysis
All quantitative assessments were carried out in quadruplicate, and the experiments were repeated 3 times. The data were expressed as mean ± SD. Statistical analysis was performed by the one-way ANOVA, using the statistical software.