Aggregation kinetics of α-crystallin
Aggregation of α-crystallin was studied under the effect of different protein concentrations.
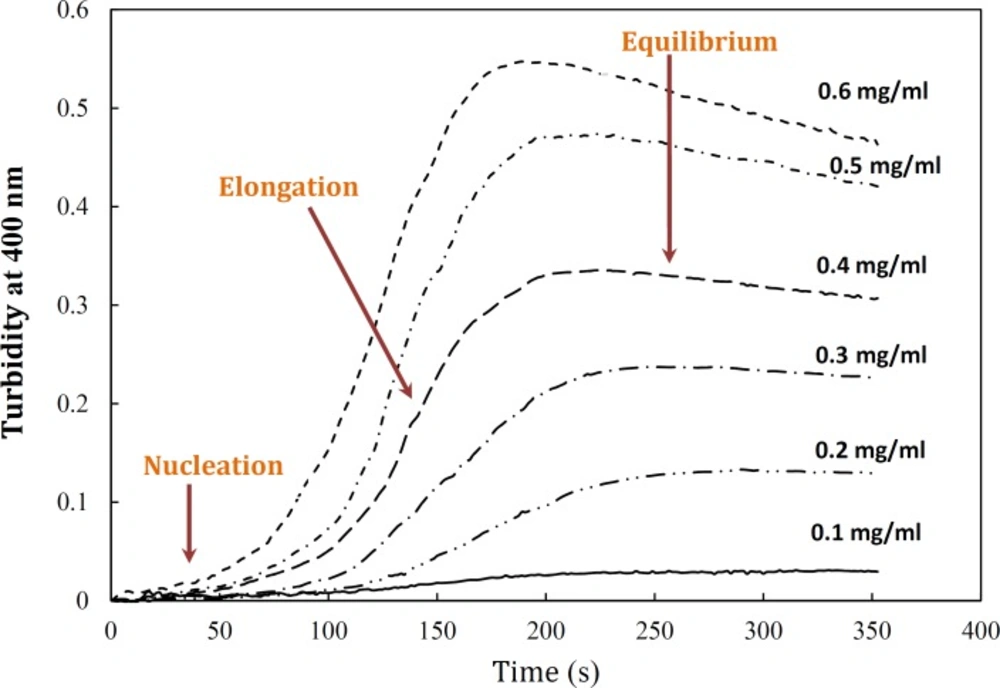
Figure 4 demonstrates a gradual development of turbidity at 400 nm with increase in α-crystallin concentration. As shown by this figure, the duration of lag time was relatively large and reciprocally dependent on [P]
o. As depicted in
Figure 4, the amyloid formation process was found to obey the characteristic nucleation – elongation pattern, with three distinct phases: initial nucleation, elongation and equilibrium.
Effect of protein concentration on the kinetics of GdnHCl-induced α-crystallin aggregation at 60 °C. The final concentrations of the protein are shown within figure. Turbidity changes were normalized according to maximal change observed. The modified crystallin displayed no detectable aggregation (data not shown). Data shown are one representative example of three independent experiments. Further details are given in experimental procedures
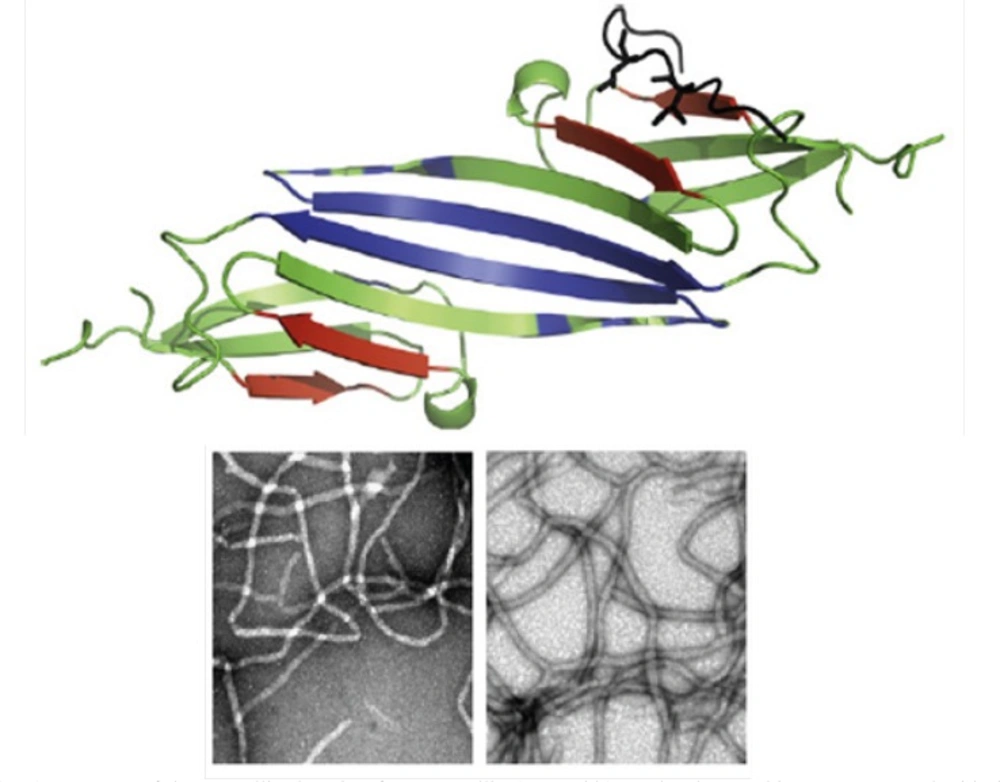
Surprisingly, the modified crystallin displayed no amyloid fibrillation under GdnHCl-induced α-crystallin aggregation conditions. It is believed that hydrophobic interactions are main driving forces of crystallin amyloid aggregation ((
21), see also
Figure 5). To re-test this possibility, PSH of the native and modified proteins were determined (
20). As indicated in
Table 1, PSH of the crystallin decreased after modification. These results are in full agreement with turbidimetric (
Figure 1) and hydropathy (
Figure 5) data confirming determinant role of hydrophobic interaction in crystalline amyloid formation.
Theoretical analyses of hydropathy profiles
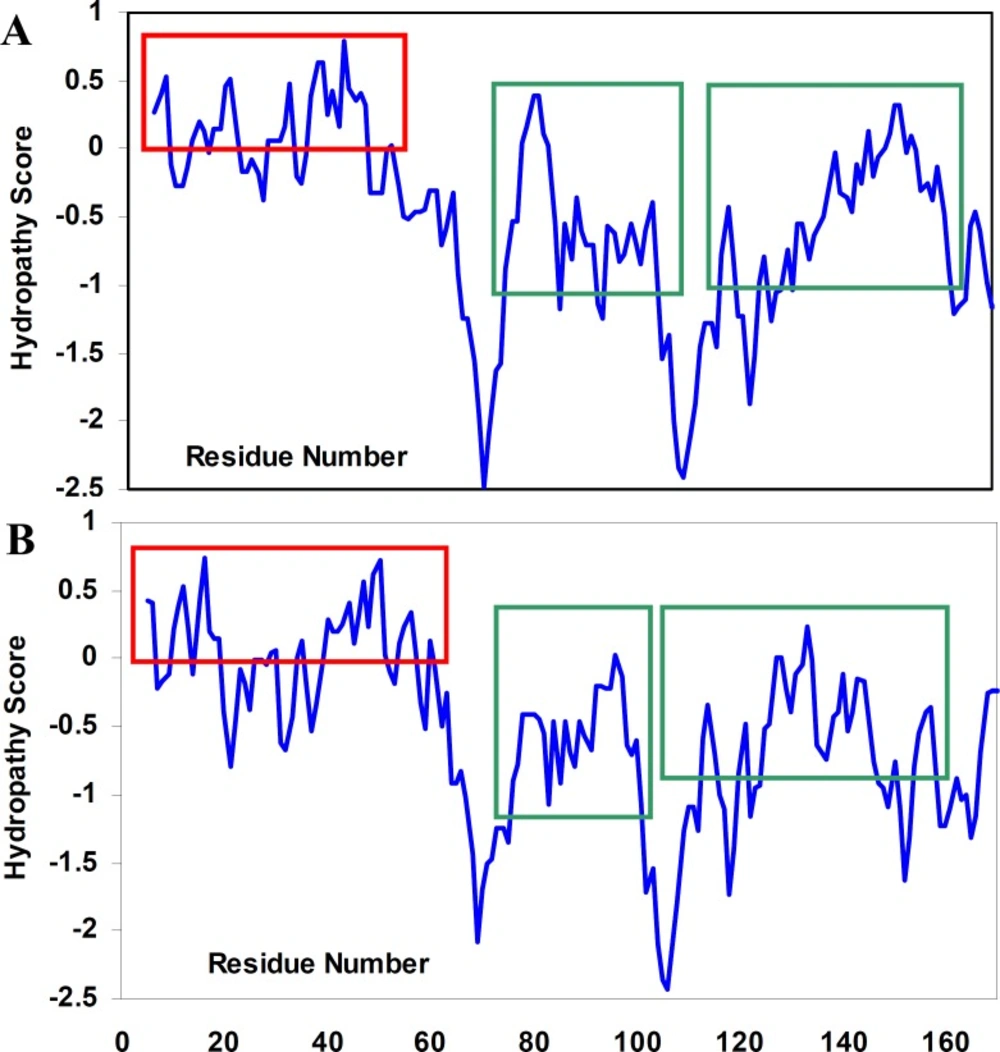
To the best of our knowledge, the structural determinants of the formation of amyloid fibrils are not clear. We, therefore, tried not only to investigate hydropathy profile in α-crystallin. Based on hydropathy profiles (
Figure 5), apolar residues of crystallins consecutively have been distributed mainly through the aggregation-prone segments of polypeptide sequences.
Hydropathy profiles of αA-crystallin (A) and αB-crystallin (B), calculated using the Roseman hydrophobicity scale (21) with window size 9. Note and compare the continuity and discontinuity in positive hydropathy scores, as indicated by red rectangulars. Also, note the homogeneity among hydropathy values in aggregation-prone segments (21) of crystallin sequences, as indicated by green rectangulars. See text for further details. For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.
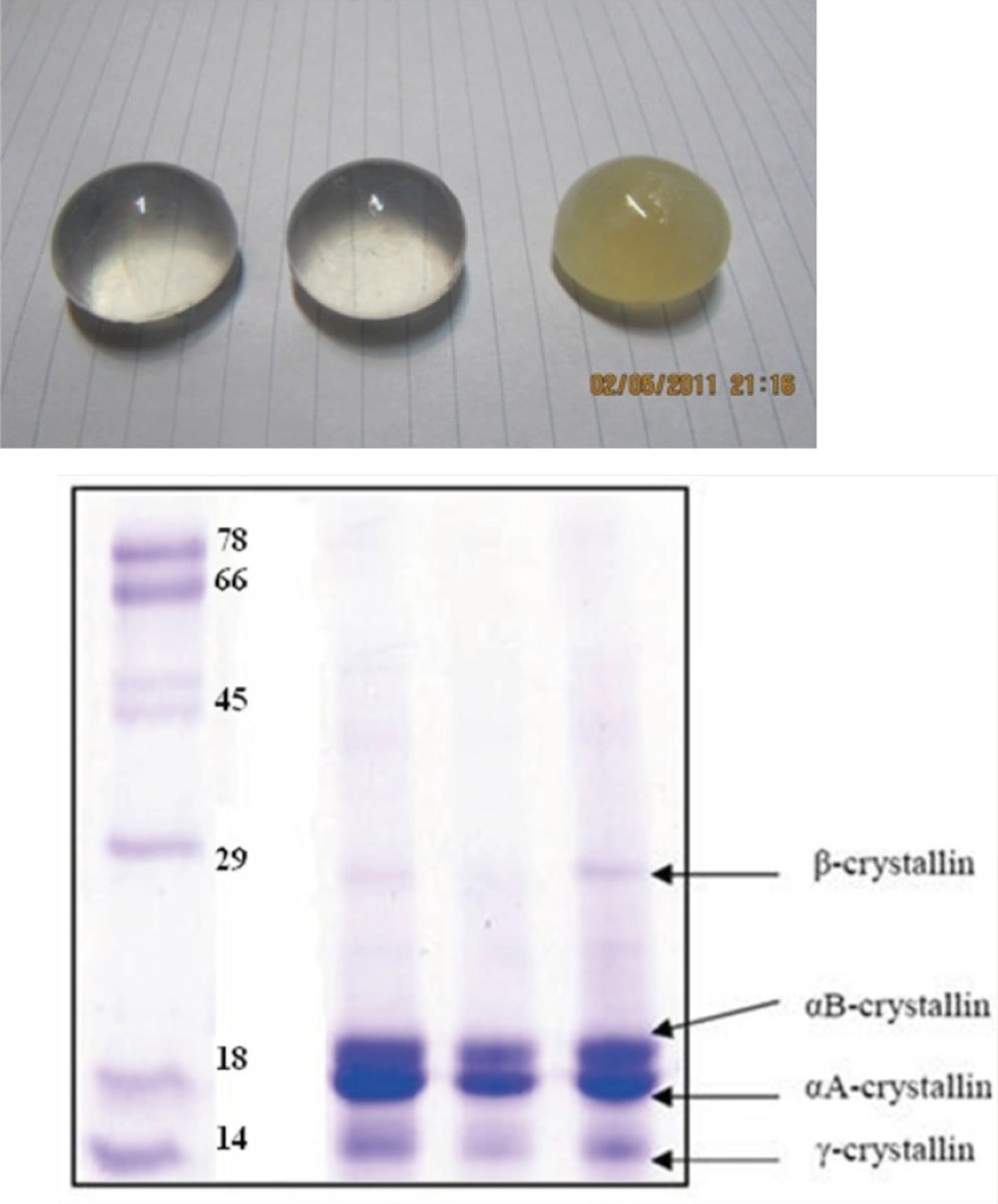
Furthermore, since the modified crystallin had no significant aggregation propensity, compared to the native protein, it was excluded from further studies.
| Ka (µM) | Y-intercept | Kd (µM) | X-intercept | [Protein] (mg/ml) | PSH |
|---|
| *Native | 0.102±0.032 | 9.80±0.23 | 48.76±2.1 | 478.1±12.1 | 0.10 | 487.6±4.5 |
| *Modified | 0.158±0.036 | 6.33±0.016 | 38.32±1.4 | 242.7±8.3 | 0.10 | 383.2±6.9 |
Data shown are mean of three independent experiments and standard deviations were within 5% of the experimental values.
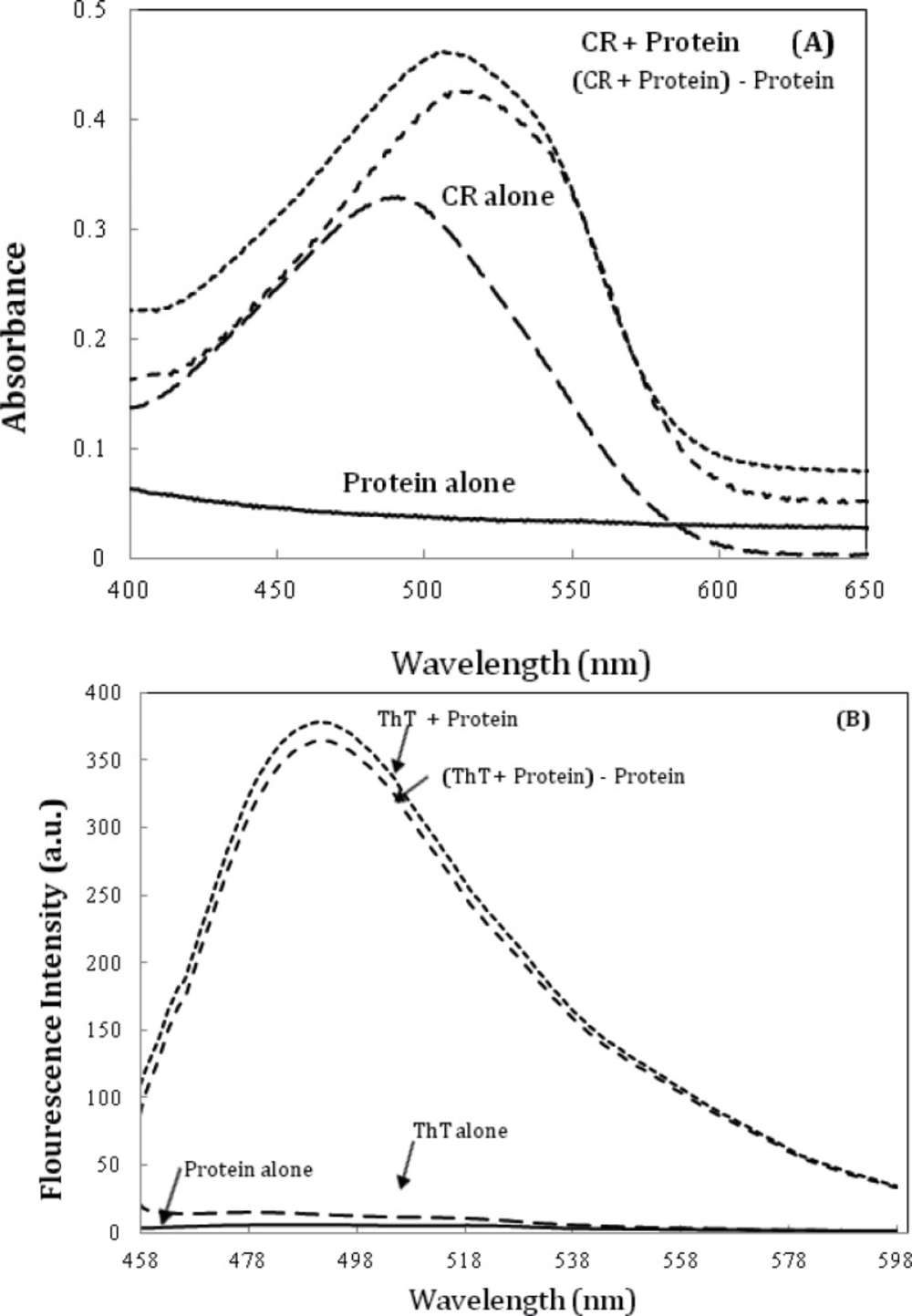
Further characterization of amyloid fibrils was performed using Congo red (CR) binding assay (
6). The absorption spectra of CR on binding to the aggregates of the α-crystallin showed a red shift of the maximum absorbance of CR from ~ 490 nm to ~ 520 nm (
Figure 6A), which is a characteristic of amyloid aggregation. The same observations were made using ThT fluorescence spectroscopy (
Figure 6B) (
6).
A) Alpha-crystallin amyloid fibrillation, as measured by monitoring changes in CR absorbance spectra. Crystallin, at 0.5 mg/mL concentration was incubated at 60 °C. The spectrum of CR alone was compared with that of CR solutions in the presence of 0.5 mg/mL Crystallin. (c) α-crystallin aggregation as indicated by ThT assay (the fluorescence intensity at 482 nm). The final protein and ThT concentrations were 0.05 mg/mL and 10 µM, respectively. Data shown are one representative example of three independent experiments. Further details are given in experimental procedures.
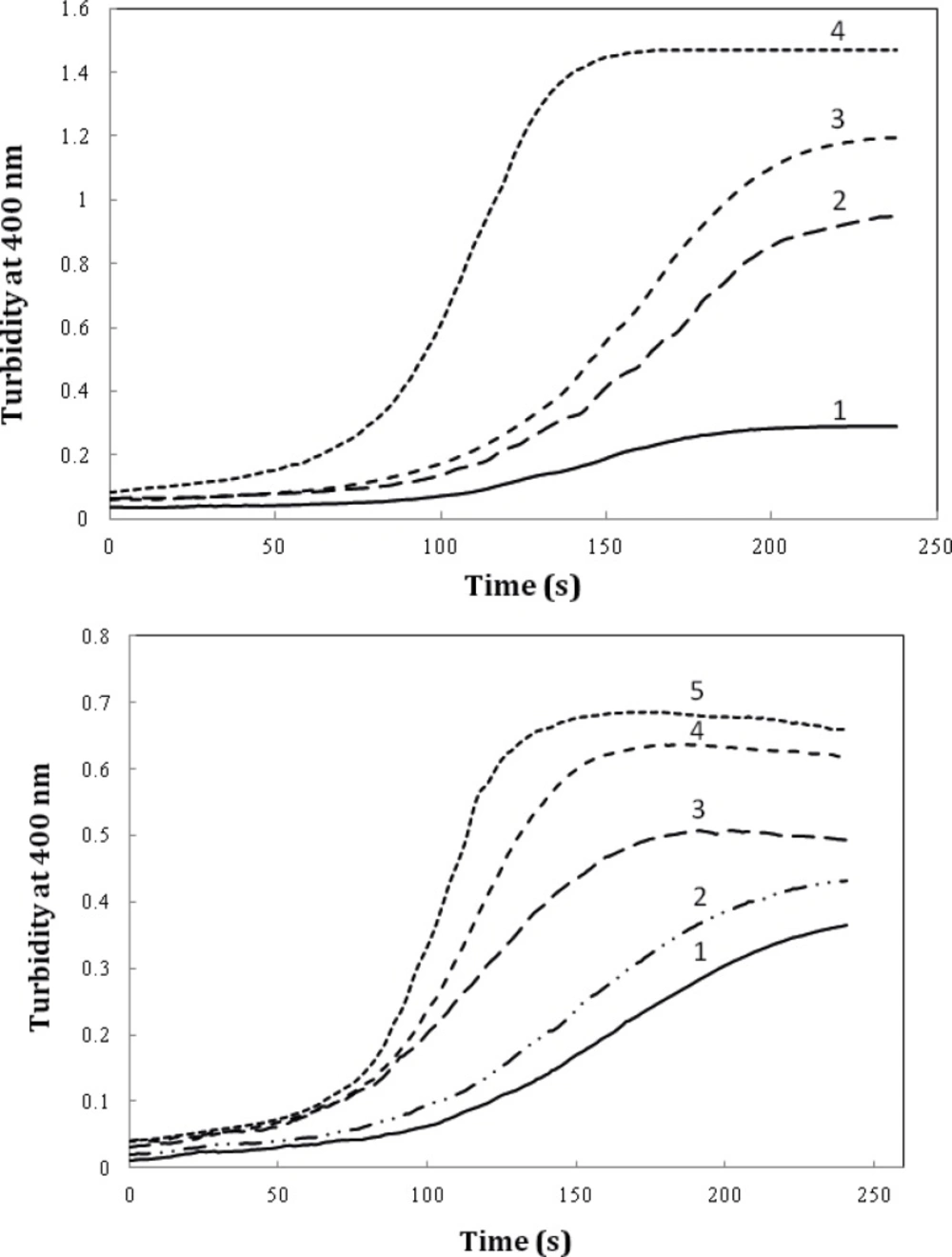
We also evaluated the effect of timolol on the extent of amyloid aggregation of crystallin. As evident from
Figure 7A, timolol (5 μM) enhances the aggregation extent of the protein, at 0.6 mg/mL, by almost ~3-fold. Also, as indicated in
Figure 7B, a significant and dose-dependent increase in the apparent aggregation extent of crystalline was observed in the presence of increasing concentrations of the drug. Further investigations are needed to disclose the mechanism of timolol-induced crystallin aggregation. Moreover, since in the present work, we have studied on Bovine α-crystallin, it is noteworthy that the human and bovine αA/αB-crystallins show more than 98% sequence identity, as revealed by amino acid sequence homology analyses.
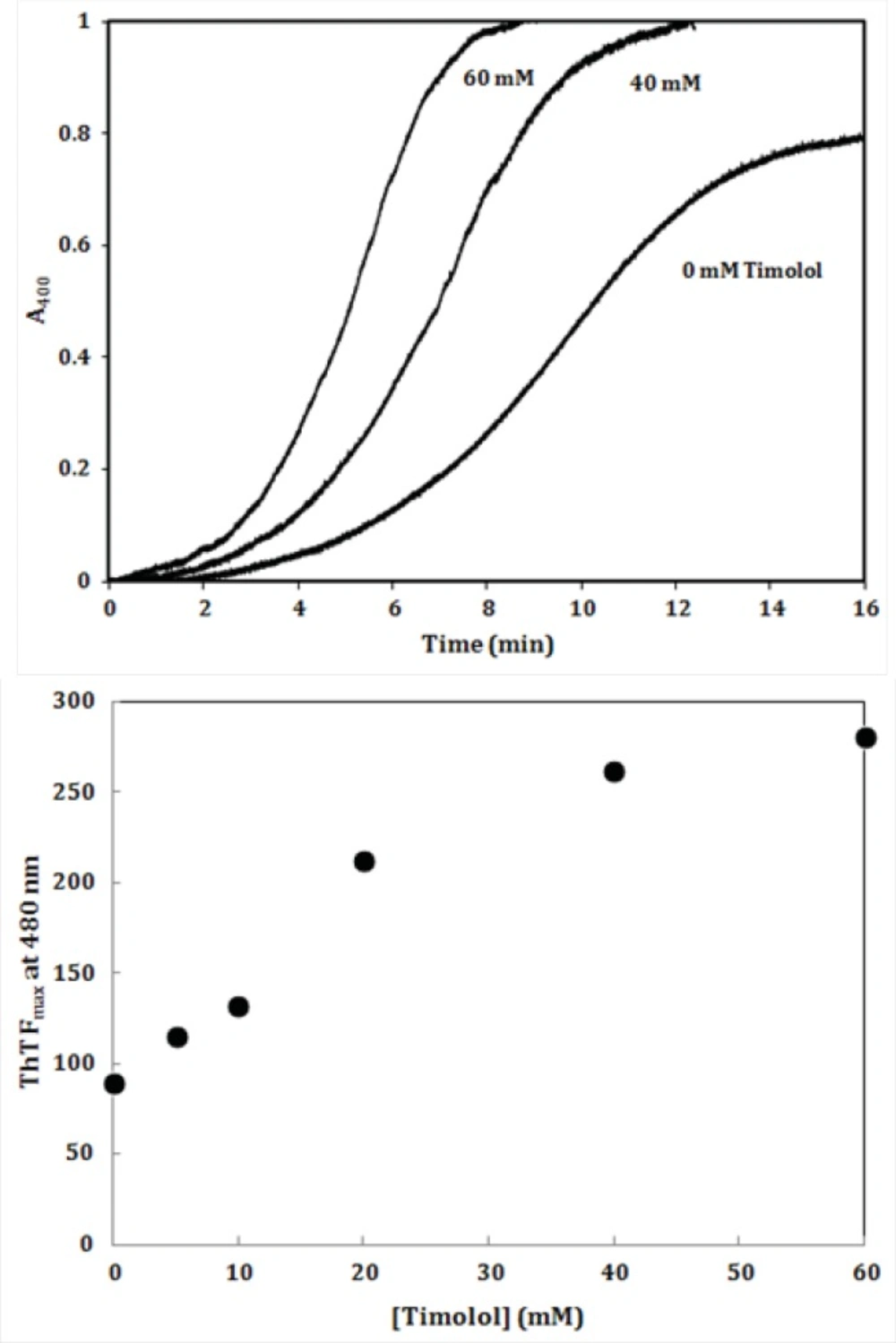
The effect of timolol on the GdnHCl-induced α-crystallin aggregation extent at 60 °C. (A) Protein (α-crystallin) and timolol concentrations were 0.6 (curves 1, 2), 0.8 (curves 3, 4) mg/mL and 5 μM, respectively. (B) After addition of α-crystallin (0.6 mg/mL) to 0.1 M sodium phosphate buffer (pH 7.4), containing 1, 2, 3, 4 and 5 µM of timolol, the absorption of each sample was measured at 400 nm with respect to the appropriate blank. Data shown are one representative example of three independent experiments
To further investigate the amyloidogenic property of timolol, heat-induced denaturation of α-crystallin was evaluated with (or without) timolol at different concentrations and the extent of aggregation was registered by monitoring the absorbance increment at 400 nm. As depicted in
Figure 8, addition of timolol to the incubation mixture induced aggregate formation of α-crystallin. At 60 mM concentration of timolol, there was more than 50% induction of the α-crystallin aggregation. Incubation of timolol alone, at different concentrations tested, also, showed no change in turbidity. We then set out whether timolol induce amyloid aggregate formation or not. To test this possibility, additional characterization of amyloid fibrils was performed using ThT fluorescence assay. The relative emission intensity of ThT (at 480 nm) on binding to the aggregates of the α-crystallin in the presence of timolol showed a significant increase from ~ 100 to ~ 250 (
Figure 8), which is a characteristic of enhanced amyloid aggregation, probably coinciding with the accelerated formation of extended β-sheet (fibril) structures. It is noteworthy that timolol has no effect on the emission spectrum of ThT.
(Top) Effect of different concentrations of timolol on the aggregation kinetics (extent/rate) of α-crystallin. The mixed solutions of α-crystallin in the heating buffer containing various concentrations of timolol were heated with the result of the turbidity development, displaying different values on the absorbance at 400 nm and the ability of timolol to induce α-crystallin aggregation was documented. (Bottom) The relative emission intensity of ThT (at 480 nm) in the presence of timolol showed a significant increase, which is characteristic of enhanced amyloid aggregation and extended β-sheet (fibril) structures. Data shown are one representative example of three independent experiments and standard deviations were within 5% of the experimental values.
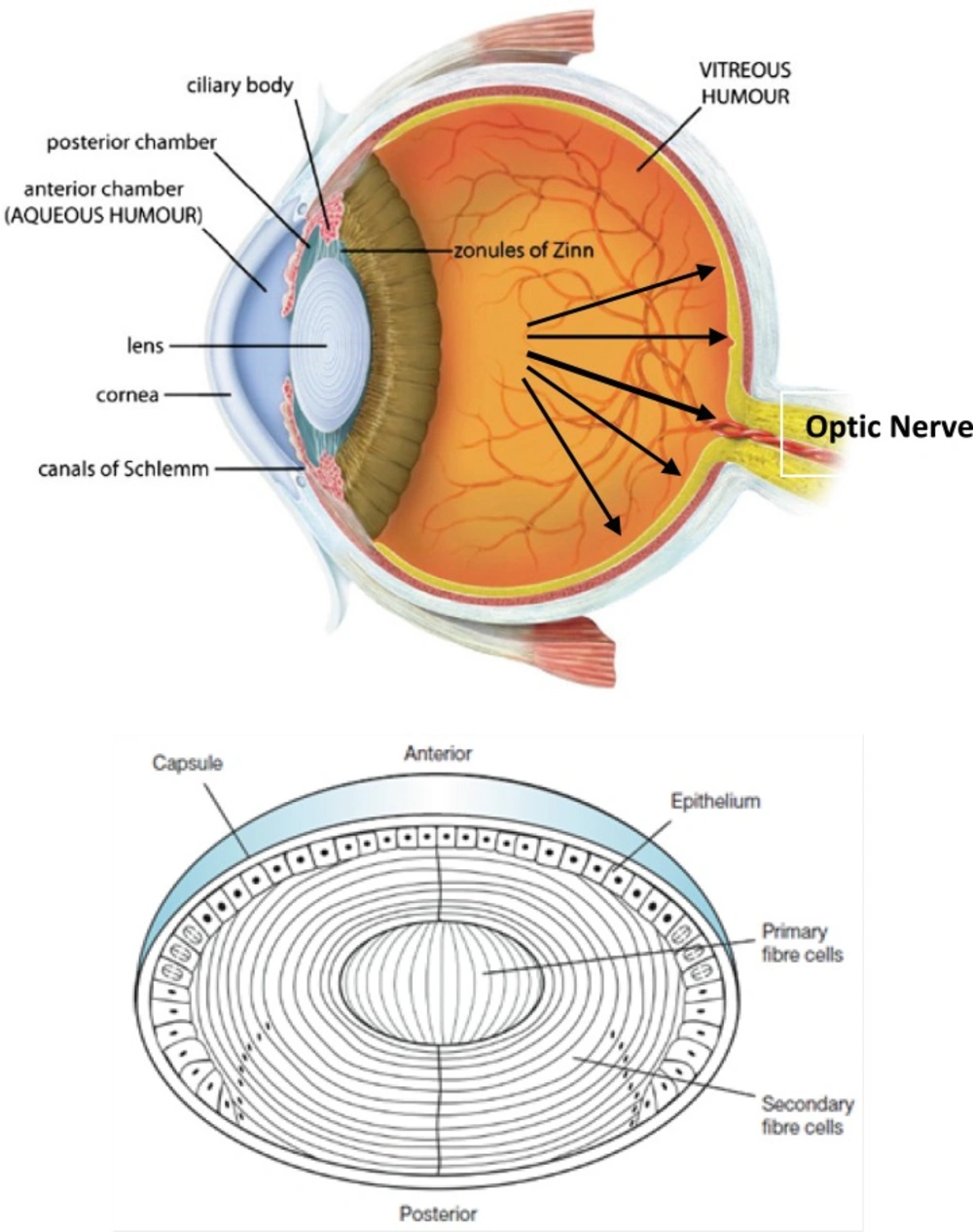
Intraocular pressure (IOP) depends on the balance between the inflow and outflow of aqueous humor. In glaucoma patients, IOP is elevated because of an increase in outflow resistance. Two aqueous humor outflow pathways exist in the eye. Aqueous mainly flows through the trabecular meshwork (TM) and Schemm`s canal (SC) to the episcleral vein, but an auxiliary uveoscleral pathway through the iris root and ciliary muscle exists, with fluid leaving the eye through the choroidal circulation or orbital tissues (
Figure 9, top, left). An increased resistance to flow in the main pathway, which carries 80% of total aqueous humor out of the eye, is predominantly responsible for elevated IOP in the many types of glaucoma in humans. In primary open-angle glaucoma (POAG), the most common type, increased outflow resistance occurs mainly in the juxtacanalicular (JCT) TM, the portion closest to SC, and in the endothelial-lined SC.
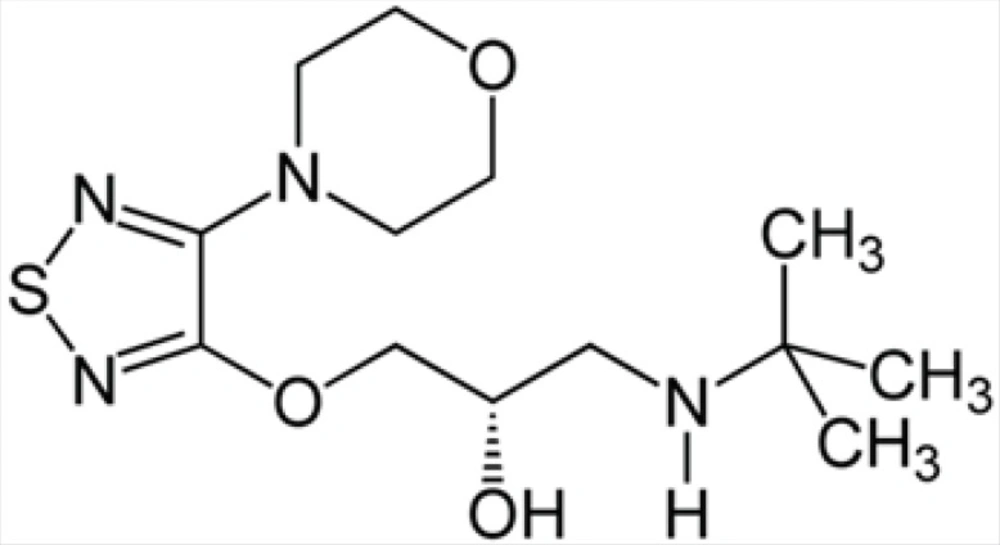
Timolol is the most frequently prescribed (non selective) β-adrenoceptor antagonist; other agents in this class include bunalol, betaxolol and carvediol which lower IOP by attenuating aqueous humor formation and enhancing trabecular outflow (
22). As a result of adrenoreceptor blockade in the ciliary body, decreased aqueous secretion, ultrafiltration, or both may occur. The most commonly reported side effects of timolol administration in people include local irritation and conjunctival hyperemia. Potential systemic risks associated with topical timolol use are related to systemic β-adrenoreceptor blockade. Cardiac arrhythmias, heart block, and bradycardia can occur with β
1-blockade, whereas pulmonary effects, such as bronchospasm and airway obstruction, can result from β
2-blockade. Beta-blocking agents are not recommended for first-line glaucoma therapy in patients with cardiovascular compromise or a history of pulmonary disease.
It is noteworthy that prescribed drugs (such as timolol) have several routes of administration and elimination from the eye which have been shown schematically in
Figure 9. (top, right) (
23).
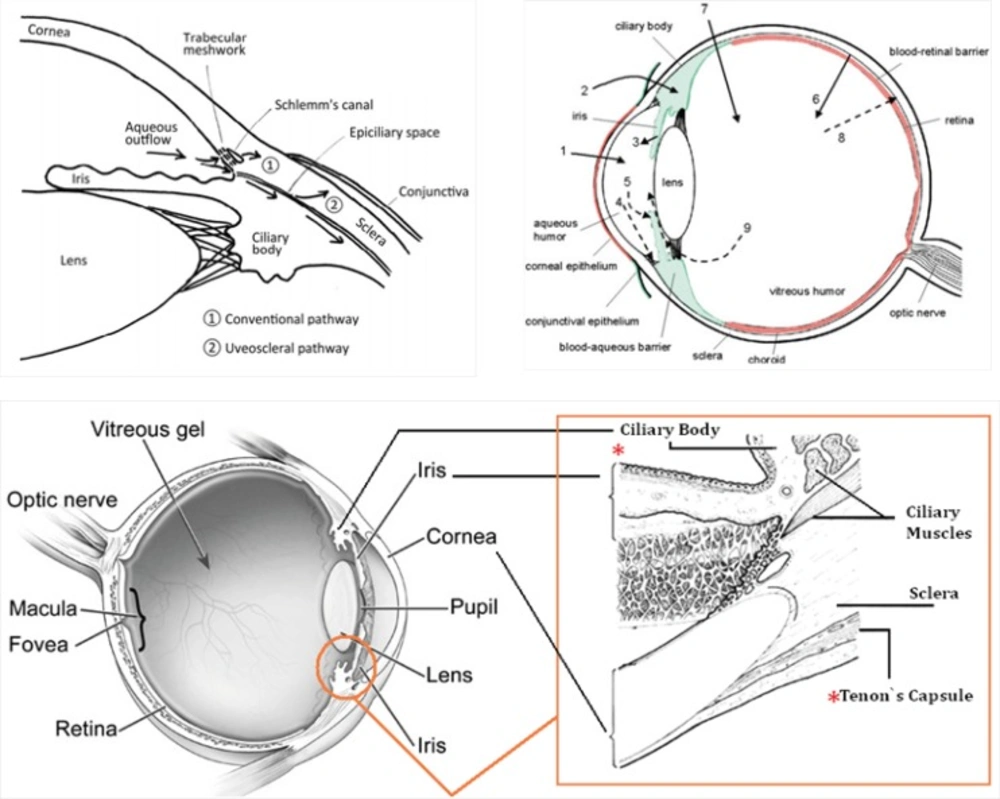
(Top, left) Schematic presentation of the two aqueous outflow pathways. Pathway is the conventional path through the TM and SC to the episcleral vein. Pathway is the uveoscleral pathway, in which aqueous travels through the iris root and ciliary muscle to the choroidal circulation or orbital tissues. (Top, right) The routes of drug kinetics. The numbers refer to following processes: 1) transcorneal permeation from the lacrimal fluid into the anterior chamber, 2) non-corneal drug permeation across the conjunctiva and sclera into the anterior uvea, 3) drug distribution from the blood stream via blood-aqueous barrier into the anterior chamber, 4) elimination of drug from the anterior chamber by the aqueous humor turnover to the trabecular meshwork and Sclemm's canal, 5) drug elimination from the aqueous humor into the systemic circulation across the blood-aqueous barrier, 6) drug distribution from the blood into the posterior eye across the blood-retina barrier, 7) intravitreal drug administration, 8) drug elimination from the vitreous via posterior route across the blood-retina barrier, and 9) drug elimination from the vitreous via anterior route to the posterior chamber. Taken from ref. (23). (Bottom) sites of action (Ciliary Body) and accumulation (Tenon’s Capsule) of timolol
Taking above statements into account and since timolol needs to penetrate within intraocular cavity to interact with its target site (ciliary body), there is this possibility that the drug diffuse into eye lens (neighbor of Ciliary Body, see
Figure 9, top, left and bottom) and interact with c
rystallins that compose over 90% of the protein within the
lens. Furthermore, periocular accumulation of timolol in glaucoma patients under long-term therapy has been previously reported so that more than 1 mg of the β-adrenergic antagonist was estimated to be present periocularly within the intact Tenon’s Capsule (
24). Thus, the drug accumulation within lens may be anticipated. At these conditions and as documented in this study, timolol-crystallin interaction may promote/trigger
in-vivo (amyloid) aggregation of α-crystallins.
In summary, since timolol and other anti-glaucoma drugs are widely administered worldwide (
25), there is the possibility that prolonged treatment of enhanced IOP play an undesired causative role in triggering onset of lens opacity, especially, in susceptible individuals carrying mutant forms of crystallin proteins. Also, the resulting data (if established) may be useful in providing mechanistic insights to develop potential curative and/or preventive strategies
in-vivo against amyloid-related cataracts. This type of aggregation-based challenges like
in-vitro protein aggregation (a serious problem in the formulation of therapeutic proteins (
26)) needs more attention.