Reagents
MTS (3-(4, 5-Dimethylthiazol-2-yl) -5-(3-carboxymethoxyphenyl) -2- (4-sulfophenyl) -2H-tetrazolium) from Promega (Madison, WI, USA); (RPMI-1640) and FCS from Gibco; Lympholyte®-H from Cedarlane (Canada); β-actin and PARP antibodies, anti- rabbit IgG and HRP linked antibody from Cell Signaling technology (Boston, USA); ECL Western blotting detection reagent from Bio-RaD (USA); the fluorescent probe propidium iodide (PI), protease inhibitor cocktail, phosphatase inhibitor cocktail, sodium citrate, Triton X-100, phenylmethylsulfonyl fluoride and QuantiPro BCA assay kit were purchased from Sigma (Steinheim, Germany).
Plant materials
The root of
R. turkestanicum was collected from Chenar, a village in Zavin Rural District, Kalat County, Razavi Khorasan Province, Iran.The plant was identified by M. R. Joharchi, from Ferdowsi University of Mashhad Herbarium. Voucher specimen (No. 42082) was deposited in Ferdowsi University of Mashhad Herbarium. Dried
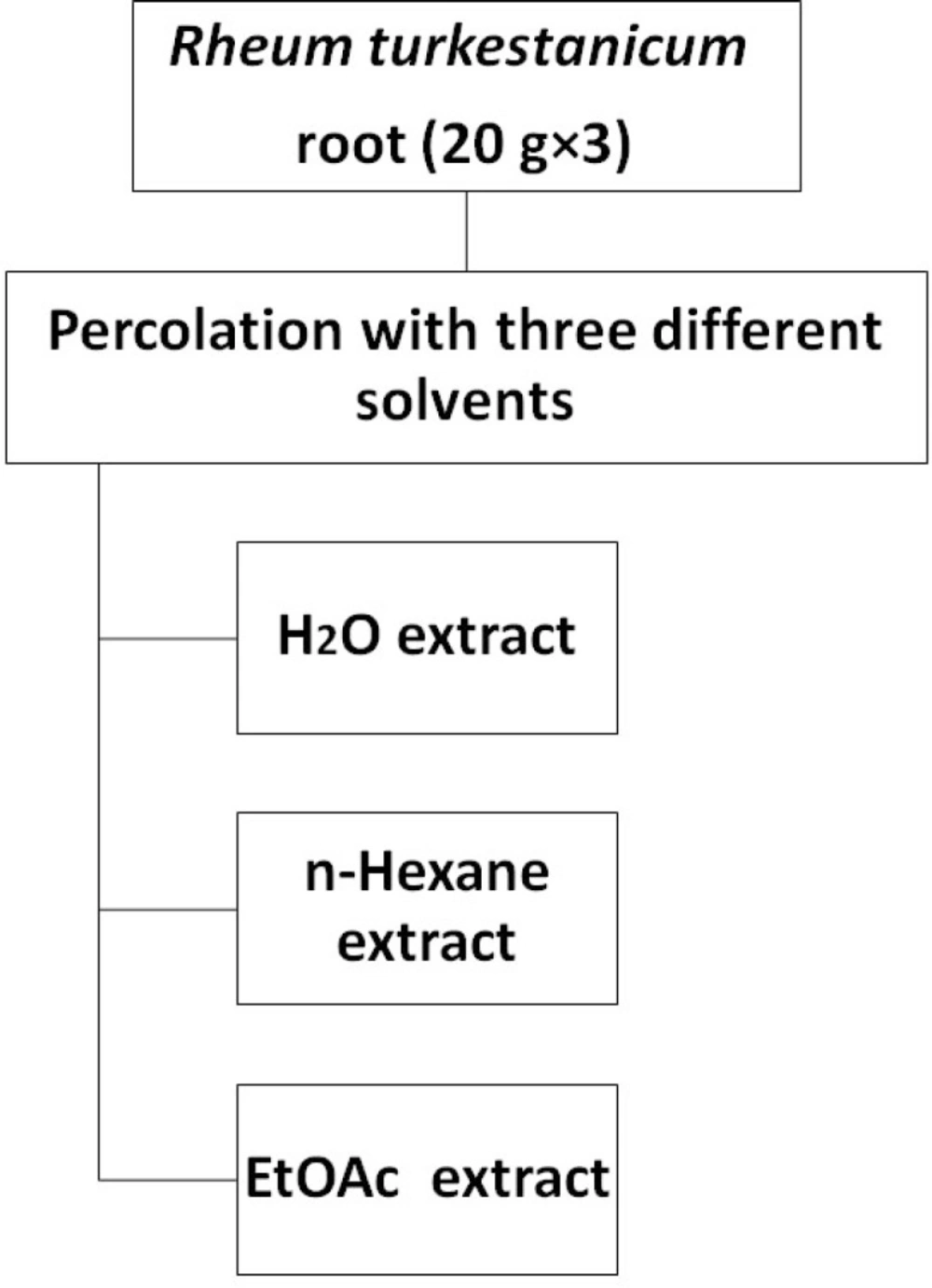
R. turkestanicum root (20 g×3) was ground into fine powder and then was percolated with 100 ml of each solvent (EtOAc,
n-hexane and H
2O). After 24 h, the solutions were centrifuged at 2700 g for 3 min. The supernatants were collected and the residues were re-extracted for one more time with the same volume of solvents (
Figure 1). The whole extract were filtered and the solvents were evaporated under reduced pressure at 40–45 °C, each extract was then stored at -20 °C over night, and then it was concentrated by freeze drier. Again the extract kept at -20 °C. The dried extrcts were dissolved in dimethylsulfoxide (DMSO) and were then screened for tumor cell growth inhibition.
Extraction scheme of R. turkestanicum
Cell culture
HeLa and MCF-7 cells were obtained from the Pasteur Institute (Tehran, Iran) and lymphocytes isolated from human peripheral blood using the Lympholyte®-H (a density gradient separation medium) according to manufacturer’s protocol and maintained at 37 °C in a humidified atmosphere (90%) containing 5% CO2. Cells were cultured in Roswell Park Memorial Institute-1640 (RPMI-1640) with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. For each concentration and time course study, there was a control sample which remained untreated and received the equal volume of medium.
Leukocyte culture
Human umbilical cord blood samples (50 mL) were collected from a fresh umbilical cord attached to the placenta by gravity flow in a sterile 50 mL syringe containing citrate buffer as an anticoagulant. The sample was diluted with an equal volume of phosphate buffered saline (PBS) and then layered over Lympholyte®-H a density gradient separation solution, and centrifuged at 800 g for 20 min at room temperature. The mononuclear cell layer was removed, washed twice in PBS and resuspended in RPMI 1640 medium supplemented with 2 mM glutamine (Sigma Chemical Co.), antibiotics and 10% FCS. Leukocytes (5×104 cells per well) were incubated with
various concentrations of R. turkestanicum in 96-well plates for 48 h. This study protocol was approved by the ethical committee of Mashhad University of Medical Sciences.
Cell viability
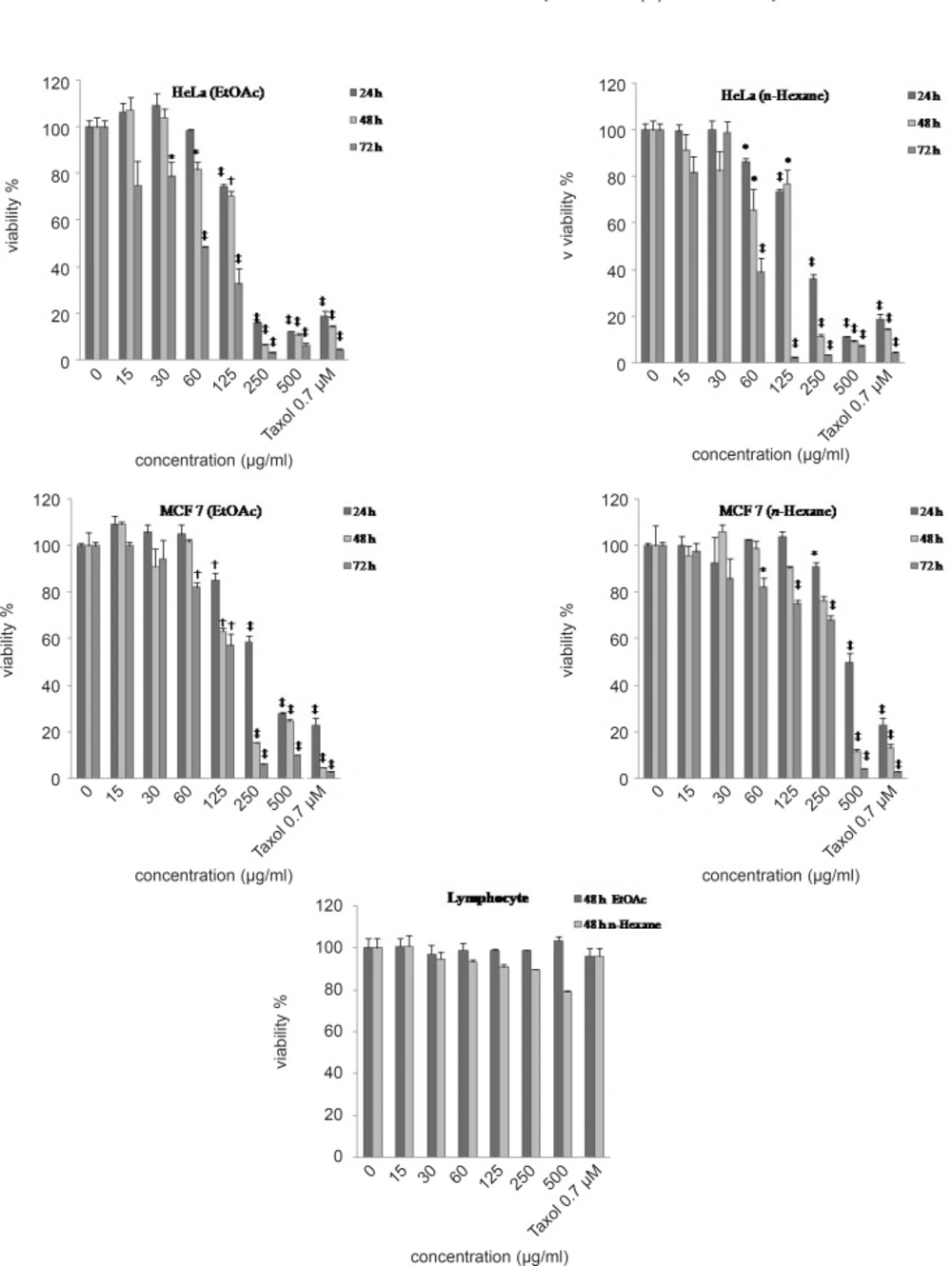
The MTS [3- (4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2- (4-sulfophenyl) -2H-tetrazolium] growth inhibition assay (
14) was performed according to the instructions provided by the manufacturer (Promega). Briefly, the cells were seeded (10
4 cell/well) onto flat-bottomed 96-well culture plates. The cells were either treated with ethyl acetate,
n-hexane and H
2O extracts (15-500 μg/mL) over different incubation periods (24, 48 and 72 h), or remained as untreated controls. At the end of each time point, fresh complete medium containing 10 μL of MTS solution was added and further incubated for 2 h. Optical density of each culture was then recorded at 490 nm using an ELISA reader. Each experiment was performed in triplicate and all the extracts were compared with Paclitaxel (0.7 μM) as a positive control. Results are expressed as the percentage growth inhibition with respect to the untreated cells.
Apoptosis
PI staining
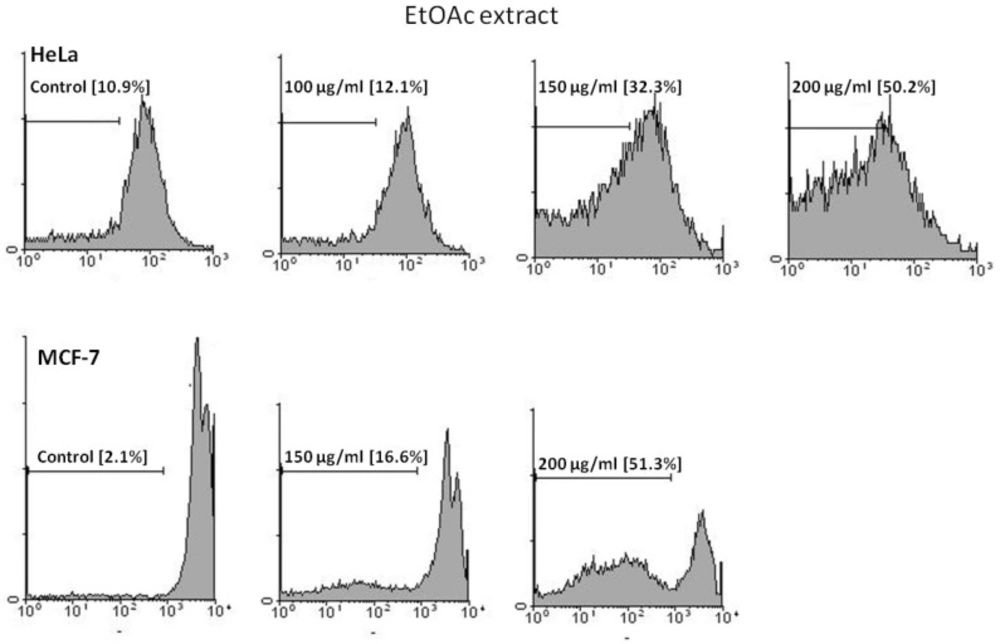
Apoptotic cells were detected using PI staining of treated cells followed by flow cytometry to detect the so-called sub-G1 peak (
15). Briefly, malignant cells were cultured overnight in a 24-well plate and treated with extracts (EtOAc and
n-hexane) for 48 h. Floating and adherent cells were then harvested and incubated at 4 °C overnight in the dark with 750 μL of a hypotonic buffer (50 μg/mL PI in 0.1% sodium citrate plus 0.1% Triton X-100) before flow cytometric analysis using a FACScan flow cytometer (Becton Dickinson). About 10,000 events were acquired with FACS.
Western blotting analysis
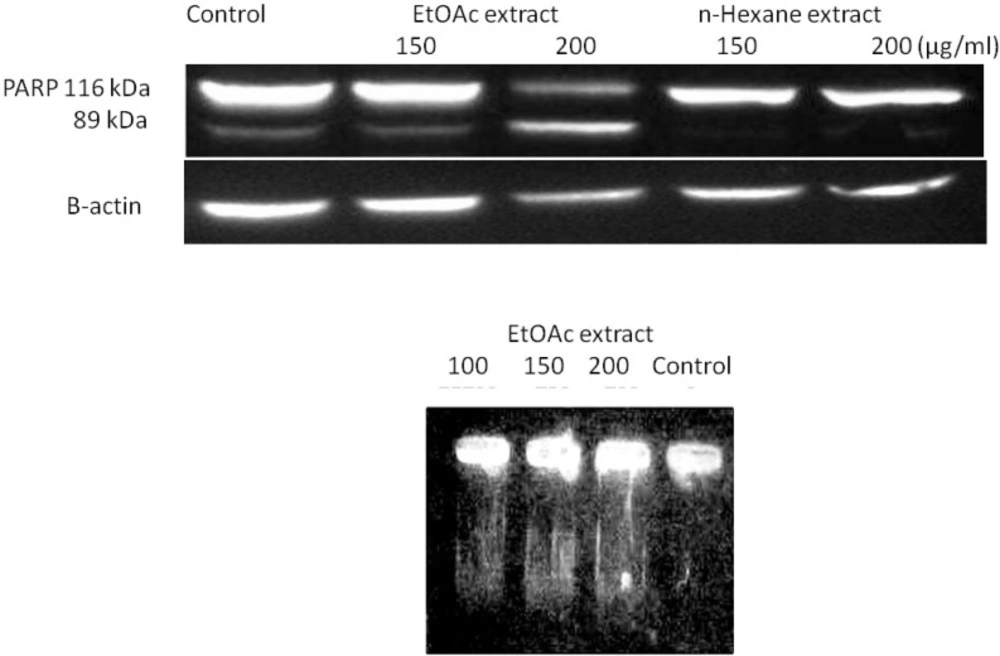
HeLa cells were treated with 150 and 200 μg/mL of the EtOAc and
n-hexane extracts of
R. turkestanicum for 48 h. The cells were harvested and rinsed with ice-cold PBS. The cell pellet was resuspended in a lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% triton-X100, 1 mM EDTA, 0.2% SDS, 1% protease inhibitor cocktail, 1% phosphatase inhibitor cocktail and 1 mM phenylmethylsulfonyl fluoride and left on ice for 30 min. After centrifugation at 10000 rpm for 20 min at 4 °C, the cell lysate was collected and protein concentration was determined according to the BCA detection kit (
16). Equal amounts of proteins were subjected to 12.5% SDS–PAGE (w/v). The proteins were transferred to a polyvinylidene fluoride (PVDF) membrane and subjected to immunoblotting using
β-actin antibody and PARP antibody as primary antibodies and anti- rabbit IgG, HRP linked antibody, as secondary antibodies. PARP cleavage in HeLa cells were detected by enhanced chemi luminescence using the ECL western blotting detection reagent.
DNA fragmentation analysis
To evaluate oligonucleosomal fragmentation, genomic DNA was extracted as previously described (
17). MCF-7 cells were treated with different concentrations of each EtOAc extract for 48 h in RPMI 1640 supplemented with 10% (v/v) fetal calf serum, 100 U/mL penicillin and 100 mg/ mLstreptomycin.
The formation of high molecular weight and oligonucleosomal DNA fragments was examined by agarose gel electrophoresis. Cells (106 cells) were seeded onto 6-well plates and treated for 48 h. The cells were collected by centrifugation at 1100 rpm for 7 min. The DNA from treated and untreated cells was extracted as explained below: cells were incubated with 50 μL of lysis buffer (20 mM Tris, 20 mM EDTA, 200 mM NaCl and 1% SDS) and 2 μL RNase A (500 μg/mL) for 15 min at 37 ºC. The cells were further incubated at 37 ºC for 15 min after adding 2.5 μL of 10 mg/mL Proteinase K, which had been preheated at 37 ºC for 30 min. The lysate was mixed with 10 mL of loading solution (30% ficoll, and 1% bromophenol blue in TBE), and then the DNA samples were separated in 2% agarose gel electrophoresis at 50 V, 3 h and visualized with ethidium bromide using Gel Documentation System (Far Gene Pouyesh, Tehran, Iran).
Statistical analysis
One way analysis of variance (ANOVA) and Bonferroni’s post hoc were used for data analysis. All results were expressed as mean±SEM and p-values below 0.05 were considered statistically significant.