Chemicals
1, 1ʼ-dimethyl-4, 4ʼ-bipyridilium dichloride (Paraquat, PQ), sulfanilamide and N-(1-naphthyl) ethylenediamine. 2HCL were purchased from Sigma-Aldrich (Germany). Trizol was obtained from Invitrogen, Life Technologies (The Netherlands). Thiobarbituric acid, phosphoric acid (85%), dimethyl sulfoxide (DMSO), ethanol and sodium nitrite were obtained from Merck (Germany). N-butanol was obtained from Carl Roth, GmbH Co. (Germany).
Experimental animals
This study was performed on 24 male and healthy Wistar rats, 10-12 weeks old, weighing between 180-200 g, kept at the animal center of Urmia University. The animals were kept in ventilated room at 22 ± 2 °C with a 12 h light/dark cycle. The rats were provided with food and water ad libitum. All performed experiments on animals were in accordance with the guidelines of the ethical committee for research on laboratory animals of Urmia University.
Experimental design
Following a week acclimation, the animals were assigned into four groups (n = 6) as control and test groups. Before the experimental procedures, all animals were weighted and these procedures were repeated at the end of study to evaluate any changes in the body weight gain (BWG). Animals in the control group received physiological saline in a volume (0.5 mL/rat) similar to the PQ solution in the test groups. The animals in test groups received PQ (dissolved in sterilized saline normal) at the three various doses, representing low (3.5 mg/kg), medium (7 mg/kg) and high doses (10 mg/kg) of the PQ subcutaneously, for 7 consecutive days (
10).
Serum preparation and tissue collection
On day 8, immediately after a light anesthesia with diethyl ether, blood samples were collected directly from the heart and left to clot at room temperature for one h, and then centrifuged at 3000 ×g for 10 min to obtain the serum. After blood collection, the animals were euthanized using CO2 gas in special device and the lung, liver, and kidney tissues were dissected immediately. The collected tissue samples were divided into two parts and the first part after washing with chilled normal saline, were snap frozen in liquid nitrogen and then were immediately stored at -70 °C for further biochemical and molecular analysis. The second part of the samples was preserved in 10% buffered formaldehyde for further histopathological and histochemical examinations.
NO measurement
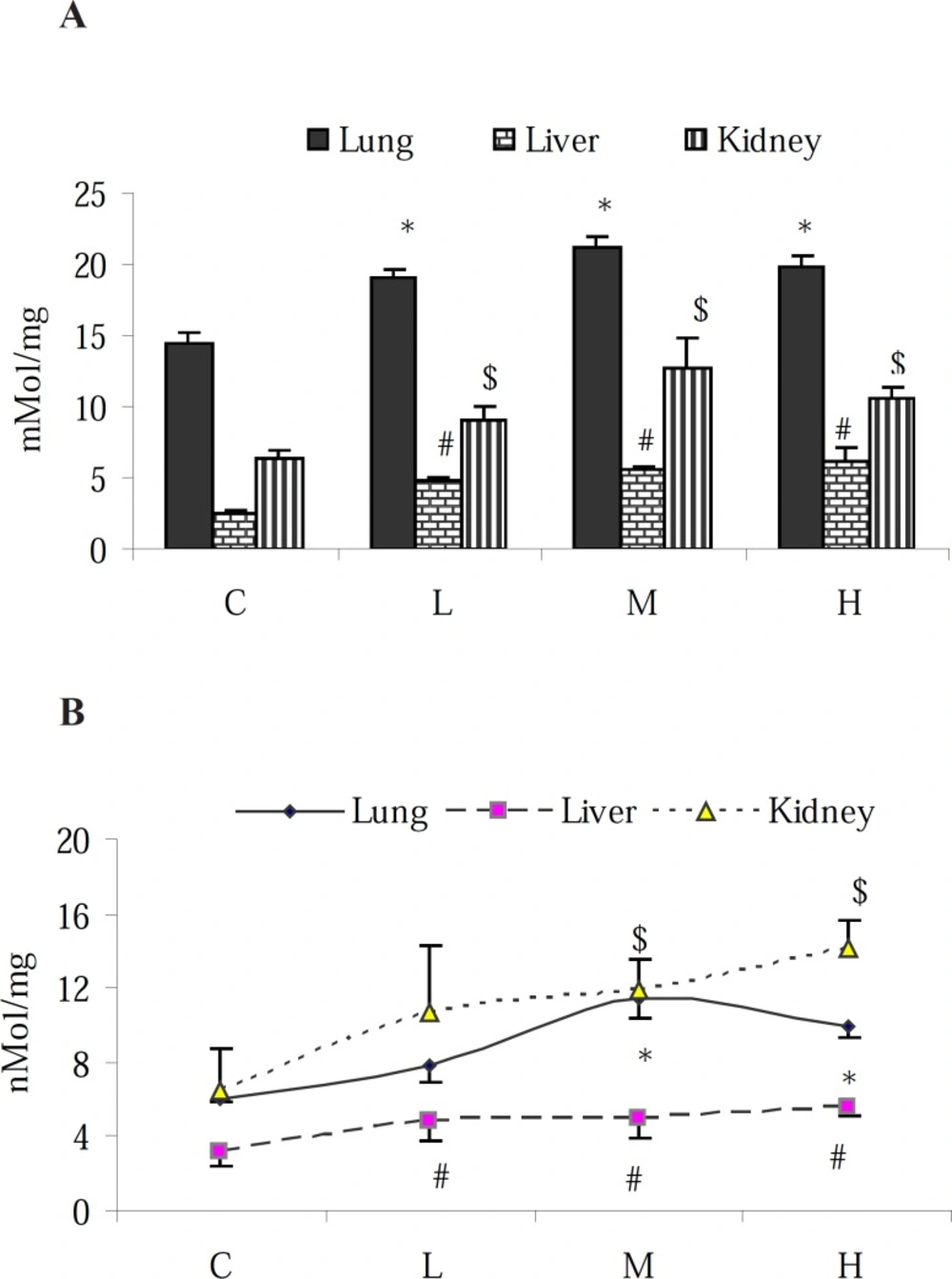
The total NO content of the lungs, liver and kidney samples was measured according to the Griess reaction (
11). In Griess reaction, nitric oxide rapidly converts into the more stable nitrite and then in an acidic environment nitrite is converted to HNO
2. In reaction with sulphanilamide, HNO
2 forms a diazonium salt, which reacts with
N-(1-naphthyl) ethylenediamine. 2HCl to form an azo dye that can be detected by absorbance at a wavelength of 540 nm. The NO content of the examined organs was expressed as nMol per mg of protein in samples.
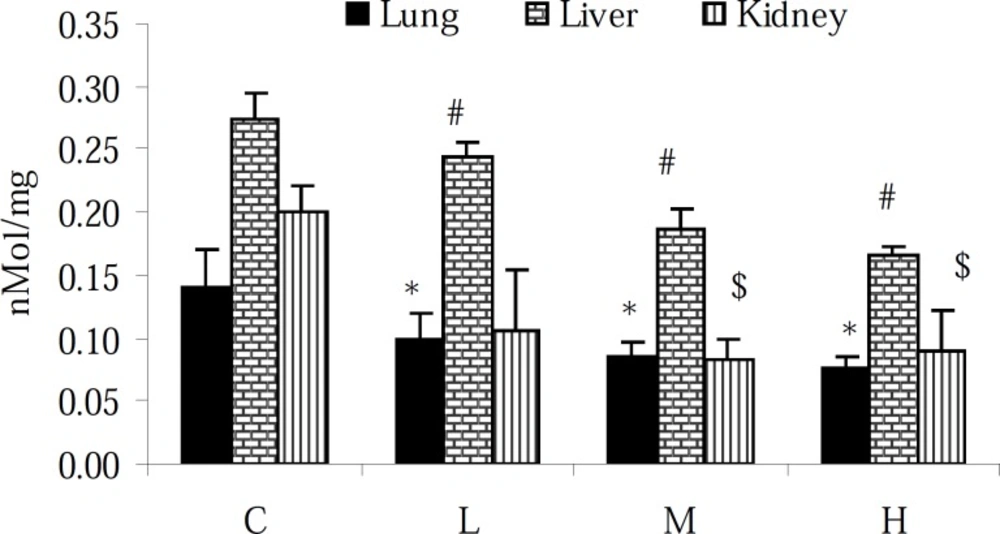
Measurement of total thiol molecules (TTM)
Total sulfhydryl levels in the lung, liver and kidney tissues were measured as described previously (
12). Briefly, 0.3-0.4 g of the tissue samples were homogenized in ice-cold KCl (150 mM), and the mixture centrifuged at 3000 × g for 10 min. Then 0.2 mL of the supernatant of the tissue homogenate was added to 0.6 ml Tris-EDTA buffer (Tris base 0.25 M, EDTA 20 mM, pH 8.2) and thereafter 40 μL l 5.5’-Dithiobis-2-nitrobenzoic acid (10 mM in pure methanol) was added to the 10 mL glass test tube. The final volume of this mixture was made up to 4.0 mL by an extra addition of methanol. After 15 min incubation at room temperature, the samples were centrifuged (Roter-Uni II, BHG, Germany), at 3000 × g for 10 min and ultimately the absorbance of the supernatant was measured at 412 nm. The TTM capacity was expressed as nmol per mg of protein in samples. The protein content of the samples was measured according to the Lowry
et al., method (
13).
Malondialdehyde (MDA) determination
To determine the lipid peroxidation rate in the control and test groups, the MDA content of the lung, liver and kidney samples was measured using the thiobarbituric acid (TBA) reaction as described previously (
14). Briefly, 0.2-0.3 g of the samples were homogenized in ice-cooled KCl (150 mM), and then the mixture was centrifuged at 3000×
g for 10 min; 0.5 mL of the supernatant was mixed with 3 mL phosphoric acid (1% V/V) and then after vortex mixing, 2 mL of 6.7 g L
−1 TBA was added to the samples. The samples were heated at 100 °C for 45 min, and then chilled on ice. After adding of 3 mL N-butanol, the samples were centrifuged at 3000×g for 10 min. The absorbance of supernatant was measured spectrophotometerically (Pharmacia, Novaspec II, Biochrom, England) at 532 nm and the amount calculated according to the simultaneously prepared calibration curve using MDA standards. The amount of MDA was expressed as nMol per mg protein. The protein content of the samples was assessed based of Lowery
et al., method (
13).
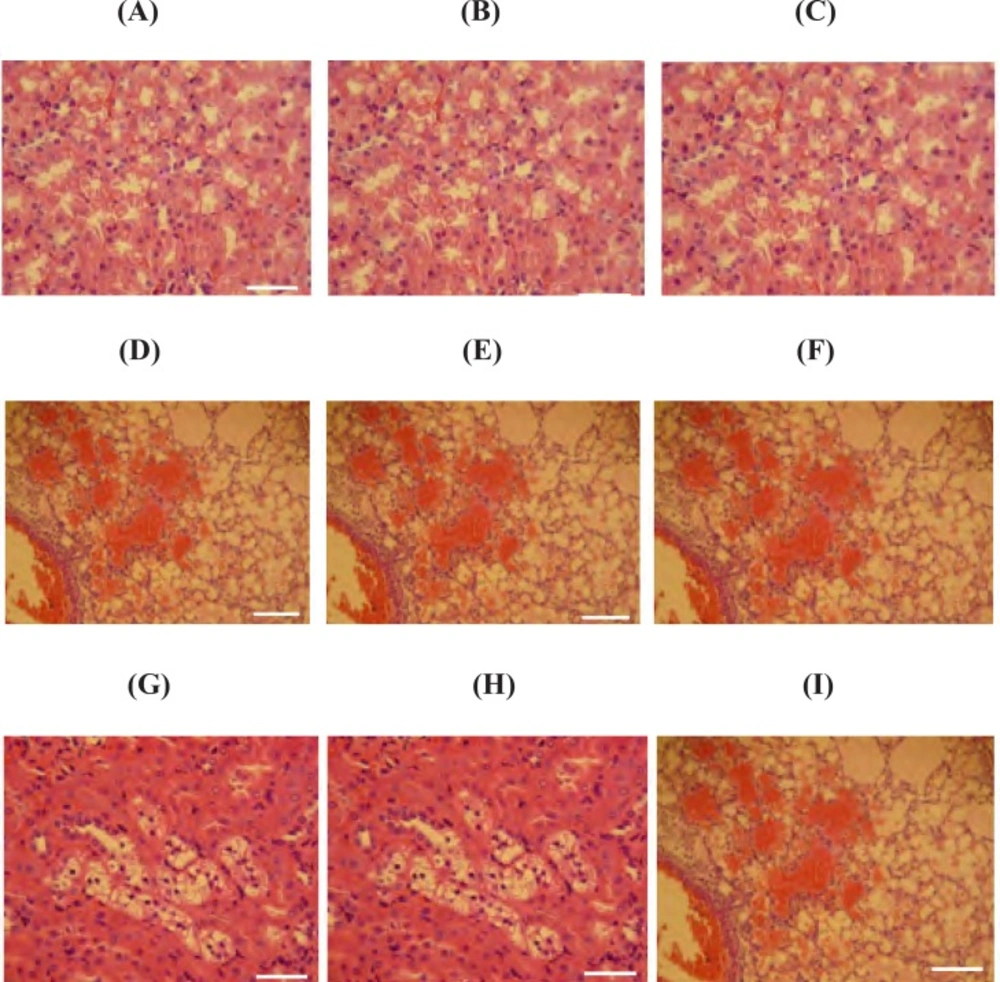
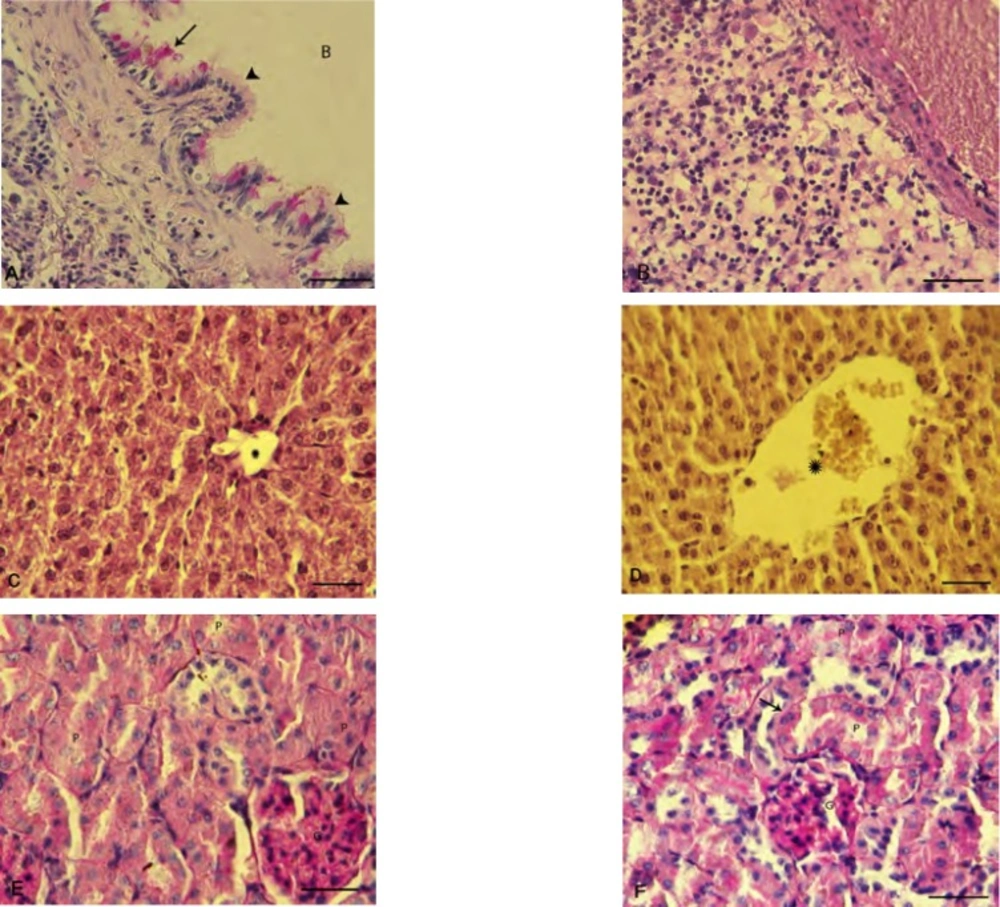
Histopathological and histochemical examinations
Tissue samples from the lungs, liver and kidneys that previously had been stored in 10% buffered formaldehyde, were embedded in paraffin and 5-6 μm sections were cut using a rotary microtome and stained with Hematoxilin & Eosin for investigation under light microscope. In order to microscopically study lesions in examined organs, special histochemical staining of periodic acid Schiff (PAS) was conducted. To evaluate the level of damages following exposure to PQ, indices such as alveolar edema and hemorrhages, hepatic glycogen degeneration and congestion and renal multifocal interstitial nephritis and presence of protein cast in renal tubules were scored numerically. The evaluation criteria were as follows: zero for no detectable lesion, 1 for mild changes, 2 for moderate changes and 3 for severe changes. For each animal in the tests and control groups, at least three slides from distinct organs were prepared and scored.
RNA isolation and RT-PCR
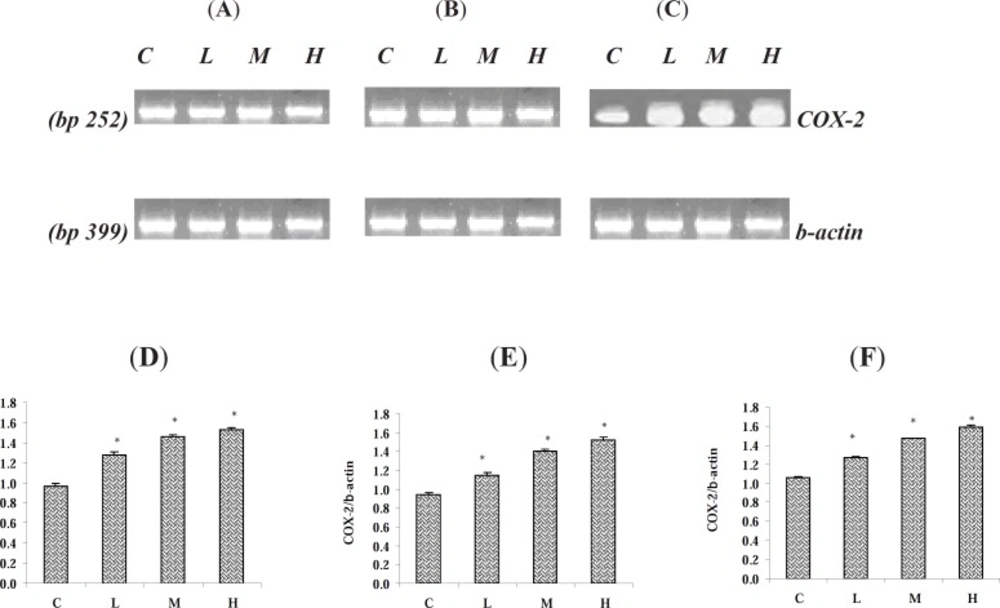
To evaluate the effect of PQ on COX-2 mRNA level, tissue samples from the lung, liver and kidney were collected and snap frozen in liquid nitrogen and then stored at -70 °C until RNA isolation. Total RNA was isolated from pooled samples homogenates for the control and test groups (n = 6) using the standard TRIZOL method (
15). To avoid genomic DNA contamination, extra care was taken when the colorless aqueous phase was collected after chloroform extraction. The RNA amount was determined spectrophotometrically (260 nm and A260/280=1.8-2.0), and the samples were stored at -70 °C. For RT-PCR, cDNA was synthesized in a 20 μL reaction mixture containing 1 μg RNA, oligo(dT) primer (1 μL), 5×reaction buffer (4 μL), RNAse inhibitor (1 μL), 10mM dNTP mix (2 μL) and M-MuLV Reverse Transcriptase (1 μL) according to the manufacturer’s protocol (Fermentas). The cycling protocol for 20 μL reaction mix was 5 min at 65 °C, followed by 60 min at 42 °C, and 5 min at 70 °C to terminate the reaction. We ran the PCR reaction with using specific RT-PCR primers –stated below- on RNA samples and no PCR bands were observed indicating that the RNA samples are free from genomic DNA.
Second strand cDNA synthesis
The RT-PCR reaction was carried out in a total volume of 25 μL containing PCR master mix (12.5 μL), FWD and REV specific primers (each 0.5 μL) and cDNA as a template (1 μL) and nuclease free water (10.5 μL). PCR conditions were run as follows: general denaturation at 95 °C for 3 min, 1 cycle, followed by 40 cycles of 95 °C for 20s; annealing temperature (62 °C for β-actin and 59 °C for COX-2) for 30 s; elongation: 72 °C for 1 min and 72 °C for 5 min. To show the lack of any genomic DNA contamination, PCR reaction was conducted on cDNA samples with two forward and reverse primers designed from intron regions of COX-2 and no PCR bands were observed on the gel, while running PCR with these two primers on genomic DNA sample extracted from rat liver, showed a 207 bp PCR fragment as we expected.
The products of RT-PCR were separated on 1.5 % agarose gels containing ethidium bromide and visualized using Gel Doc 2000 system (Bio-Rad). The density of RT-PCR bands were quantified by using the software of Molecular Analyst (Bio-Rad, the Netherlands) and normalized based on the density of corresponding
β-actin bands. The RT-PCR reaction and subsequent electrophoresis were performed three times and the averages of numerical densitometric values along with standard deviation were calculated. The specific primers for Ratus COX-2 and
β-actin were designed (
16) and manufactured by CinnaGen (CinnaGen Co. Tehran, Iran).
Primers pairs for RT-PCR were as follow:
COX-2 (the expected PCR product size = 252 bp):
Forward primer 5ʼ-TGGTGCCGGGTCTGATGATG-3ʼ and
Reverse primer 5’-GCAATGCGGTTCTGATACTG-3’,
β-actin (The expected PCR product size = 399 bp):
Forward primer 5′-CTGACCGAGCGTGGCTACAG-3′ and
Reverse primer 5′-GGTGCTAGGAGCCAGGGCAG-3′).
Statistical analysis
The mean and standard deviations of measured parameters were calculated. The results of three independent experiments for each assessment were analyzed using Graph Pad Prism software (version 2.01; Graph Pad software Inc. San Diego, California). The comparisons between groups were made by the analysis of variance (ANOVA) followed by Bonferroni post hoc test. For comparing the graded degree of pathological findings between groups, the Kruskal-Wallis test was used. A p-value < 0.05 was considered significant.