Chemicals
All the chemicals used were of analytical grade. Sodium azide (NaN3), 2-deoxyglucose (2-DG), gallic acid, potassium peroxodisulfate, 2,2’-azinobis-(3-ethyl-benzothiazoline-6-sulfonic acid (ABTS), 1,1-diphenyl-2-picrylhydrazyl free radical (DPPH), 2,4,6-tri(2-pyridyl)-s-triazine (TPTZ), nerve growth factor (NGF), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), trypan blue, Dulbecco’s phosphate buffer saline (DPBS) and other chemicals of highest purity, and cell culture media, serum and buffer constituents were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). 6-hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Trolox) was purchased from Acros Organics (New Jersy, USA). Anhydrous sodium carbonate, ferric chloride hexahydrate (FeCl3•6H2O), Folin-Ciocalteu phenol reagent, hydrochloric acid, glacial acetic acid, methanol and sodium acetate trihydrate were purchased from Merck (Darmstadt, Germany).
Composition of calcium krebs ringer (CaKR) buffer
The CaKR buffer (pH 7.4) used for ischemic cells was devoid of glucose while the one used for control cells contained 6 mM glucose. The modified CaKR buffer (without glucose) composed of 125 mM NaCl, 5 mM KCl, 2.5 mM HEPES-NaOH, 5 mM NaHCO3, 1.2 mM MgSO4, 1.2 mM KH2PO4, 1 mM CaCl2 mM, supplemented with 5 mM sodium azide and 2 mM 2-DG.
Plant material
F. arabica was purchased from Innocon Foods Pvt. Ltd., Pune, India (Batch number: C/6473). Multiple solvents (methanol: isopropyl alcohol: acetone) were used by the manufacturer for the preparation of the extract. As described, the sample (herbal extract) was authenticated for their correct botanical identity. 100 mg of the brownish colored thick gel of the extract was dissolved in 10 mL of CaKR buffer and the suspension was shaken vigorously on a vortex mixer. The suspension was kept overnight at 4°C and decanted to remove the soluble supernatant, which was filtered through a 0.22 μ syringe filter. The filtrate was collected and used as a stock (10 mg/mL) for further experimentation.
Cell culture, differentiation and determination of cell viability
Rat pheochromocytoma cells (PC12) were purchased from NCCS Pune, India, and maintained in F-12 HAM, Kaighn’s modification (HAM’s F12K) medium supplemented with 15% donor’s horse serum, 3% fetal calf serum (FCS) and 1% antibiotic antimicotic solution (Himedia, India), containing 10,000 units Penicillin, 10 mg streptomycin and 25 μg Amphotericin B/mL of culture medium. The cells were routinely incubated in 50 mL, poly-L-lysine (molecular weight > 70,000 D) coated, tissue culture flasks in humidified atmosphere of 5% CO2 and 95% air at 37°C. For experimentation 2x104 and 6x104 cells were seeded in 24 well tissue culture plates (lysine coated) for ATP and MTT respectively, while 2 × 106 cells were used for all other assays. 50 ng/mL NGF was added to the well containing differentiation media and kept for 4 days to differentiate the PC12 cells from synaptic like neurons. Differentiation media was changed on every second day. Cell viability was determined through trypan blue dye exclusion and > 95% cell viability was considered optimum for each treatment.
Induction of chemical ischemia and treatment of herbal extract
Sodium azide (5.0 mM) and 2-DG (2.0 mM) were used to induce chemical hypoxia and hypoglycemia, respectively. The chemical ischemia was induced in PC12 cells by both metabolic inhibitors according to procedures described previously (
24,
25). Once the monolayer was formed, the incubation media was removed and ischemic medium (CaKR buffer containing sodium azide and 2-DG) was introduced to cells for 2 h. Thereafter, this ischemic medium was removed and cells were washed twice with normal CaKR buffer followed by the addition of normal culture medium for 24 h, in the absence or presence of 10, 50 or 100 μg/0.2 mL concentration of herbal extract.
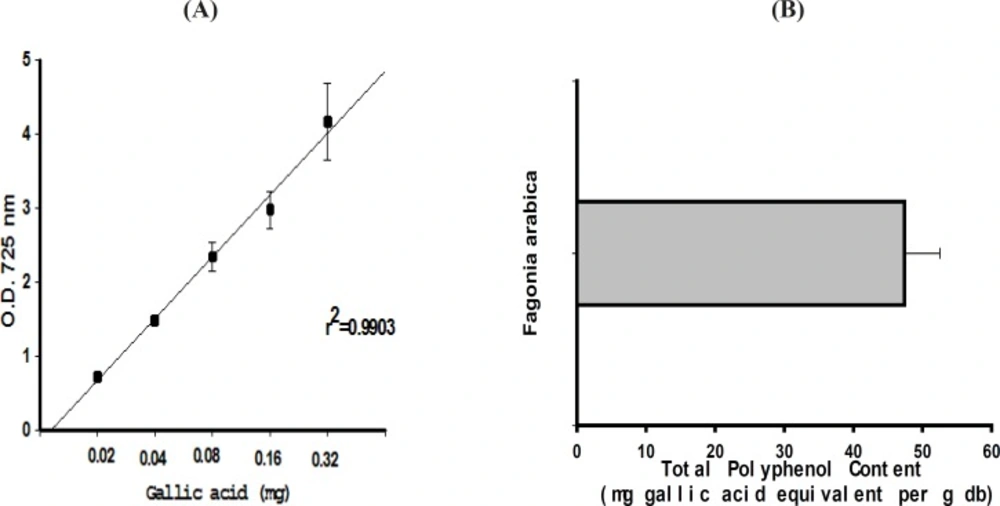
Determination of total polyphenol content (TPC)
The TPC was measured using the method described by Wong
et al., (
26). An aliquot of 100 μL of cell extract was mixed with 2.5 mL of Folin-Ciocalteu phenol reagent (10X dilutions) and allowed to react for 5 min. Then 2.5 mL of saturated Na2CO3 solution was added and allowed to stand for 1 hr and absorbance of the reaction mixture was measured at 725 nm on Helios α spectrophotometer (Thermo Electron Corpn., USA) and TPC of the extract was expressed as mg gallic acid equivalents/ g dry weight of plant.
Determination of antioxidant activity
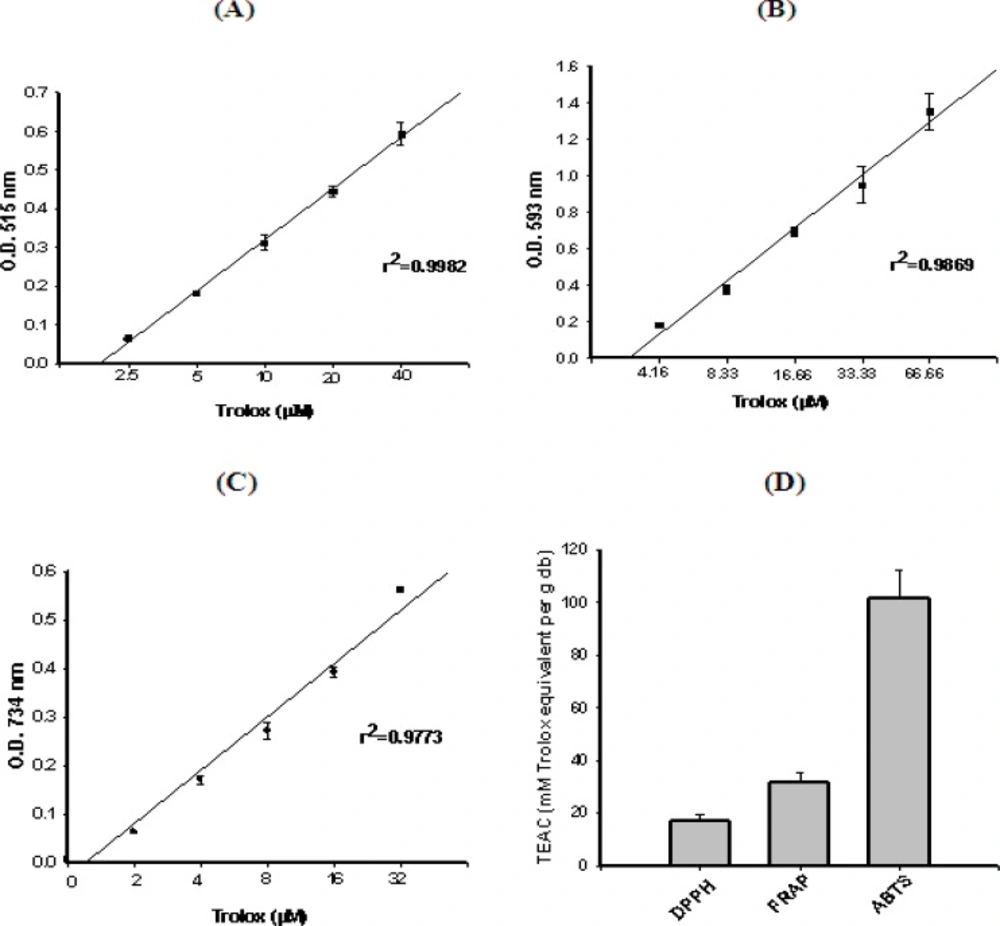
DPPH free radicals scavenging assay
The DPPH free radical scavenging activity of each sample was determined colorimetrically, according to the method described by Leong and Shui (
27). Briefly, a 0.1 mM solution of DPPH in methanol was prepared. The initial absorbance of the DPPH in methanol was measured at 515 nm and did not change throughout the period of assay. An aliquot (40 μL) of an extract (with appropriate dilution, if necessary) was added to 3 mL of methanolic DPPH solution. The change in absorbance at 515 nm was measured after 30 min. The antioxidant capacity based on the DPPH free radical scavenging ability of the extract was expressed as μM Trolox equivalents/ g dry weight of plant.
Ferric reducing antioxidant potential (FRAP) assay
The ability to reduce ferric ions was measured using a modified version of the method described by Benzie and Strain (
28). An aliquot (200 μL) of an extract (with appropriate dilution, if necessary) was added to 3 mL of FRAP reagent (10 parts of 300 mM sodium acetate buffer at pH 3.6, 1 part of 10 mM TPTZ solution and 1 part of 20 mM FeCl3•6H2O solution) and the reaction mixture was incubated in a water bath at 37°C. The increase in absorbance at 593 nm was measured after 30 min. The antioxidant capacity based on the ability to reduce ferric ions of the extract was expressed as μM Trolox equivalents/ g dry weight of plant.
ABTS free radicals scavenging assay
The antioxidant capacity assay was carried out using the improved ABTS•+ method, as described by Re
et al., (
29). Briefly, ABTS•+ radical cation is generated by reacting 7 mM ABTS and 2.45 mM potassium peroxodisulfate via incubation at room temperature (23°C) in the dark for 12-16 h. The ABTS•+ solution was diluted with 80% HPLC-grade ethanol to an absorbance of 0.700 ± 0.040 at 734 nm and equilibrated at 30°C. Plant extract was diluted with distilled water or 80% methanol, so that after introduction of a 30 μL aliquot of each dilution into the assay, it produced from 20% to 80% inhibition of the blank absorbance. To 3 mL of diluted ABTS•+, 30 μL of the plant extract solution was added and mixed thoroughly. The reaction mixture was allowed to stand at room temperature for 6 min and the absorbance was recorded immediately at 734 nm. Trolox standard solutions (concentrations from 0 to 32 μM) in 80% ethanol were prepared and assayed using the same conditions. Appropriate solvent blanks were run in each assay. The percent of inhibition of absorbance at 734 nm was calculated and plotted as a function of concentration of trolox for the standard reference data. The absorbance of the resulting oxidized solution was compared to that of the calibrated trolox standard. Results were expressed in terms of trolox equivalent antioxidant capacity (TEAC, μM trolox equivalents/g dry weight of plant) as described elsewhere (
29).
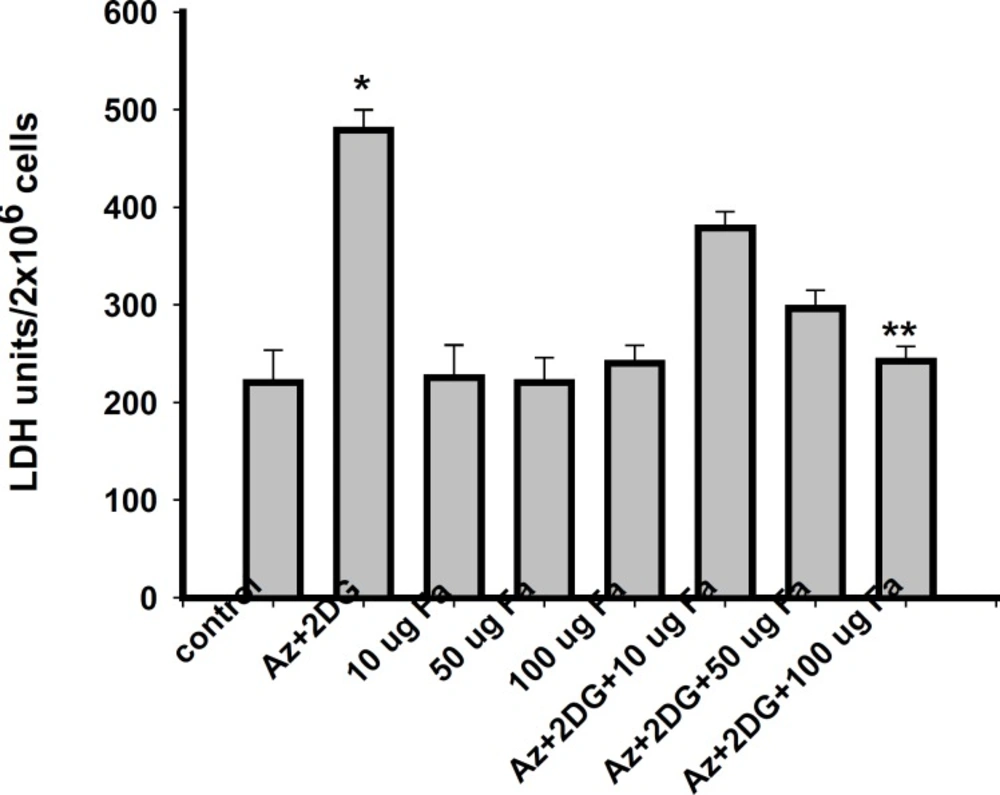
Lactate dehydrogenase (LDH) leakage assay
After reperfusion, the reperfusion media was collected and centrifuged at 2000 rpm for 10 min. The supernatant was collected and the activity of leaked intracellular LDH was measured using a diagnostic kit (Merck India Ltd., Mumbai). Briefly, LDH activity was measured at 340 nm using spectrophotometer, on the basis of reduction of pyruvate to lactate in the presence of NADH, and the values expressed as Units/2 × 106 cells (
30).
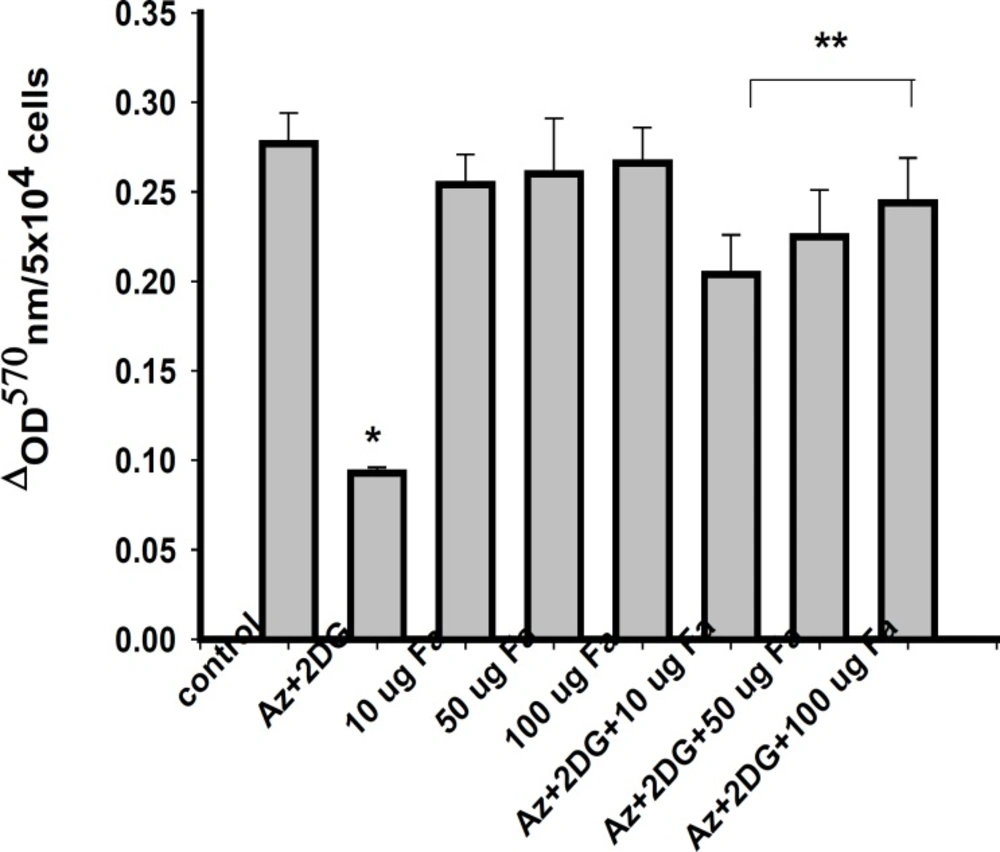
Measurement of cellular viability
Cellular viability of the cells was measured by MTT assay (
31). Briefly, 10 μL of MTT was added from the stock of 5 mg/mL to 200 μL of cell suspension, followed by 4 h incubation at 37°C in CO2 incubator. Soon after, the formazon crystals formed, were pelleted out by centrifugation and dissolved in 100 μL of DMSO and the colour developed was measured spectrophotometrically at a wavelength of 570 nm and the values expressed as
ΔOD 570/6x104 cells.
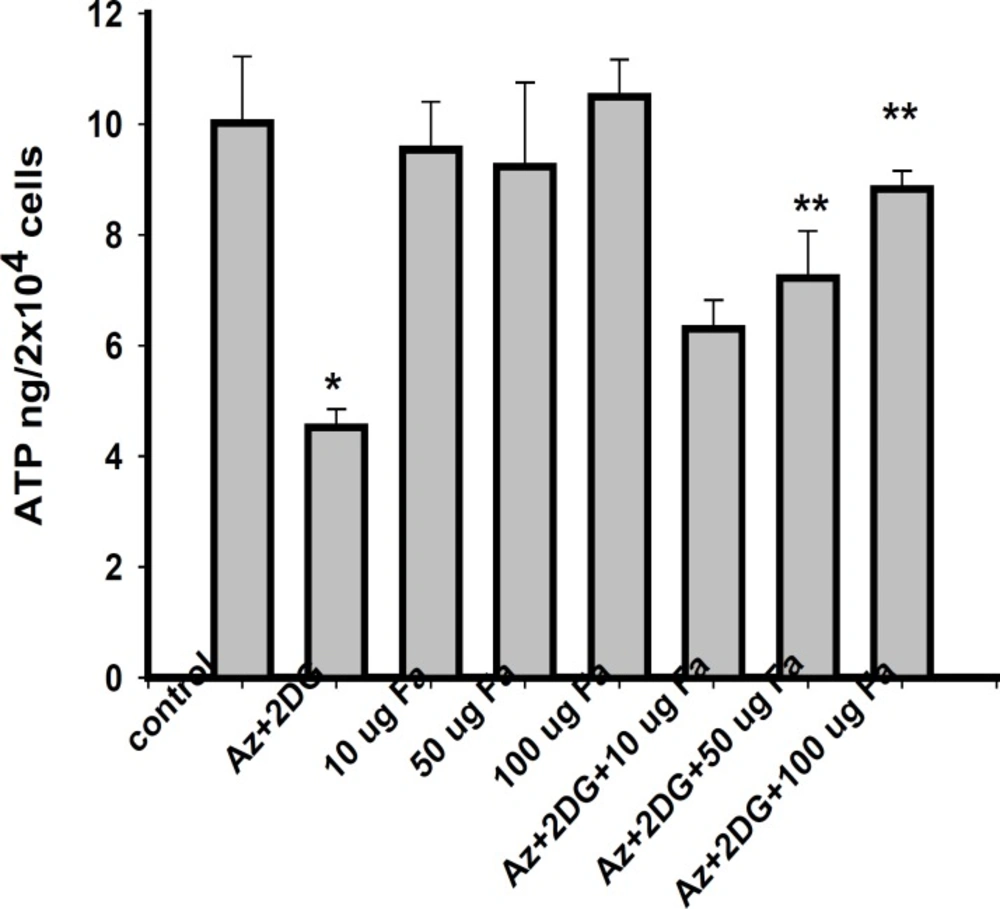
Measurement of cellular ATP
After reperfusion, ATP level in the cells was measured by ATP assay kit from Calbiochem (Germany). This kit utilizes the bioluminescence for the rapid detection of the ATP levels. The enzyme luciferase catalyzes the formation of light from ATP and luciferin. The light produced was measured on a plate reader at 562 nm. Standard curve was prepared using different concentrations of ATP (0.1 ng to 100.0 ng) and the values expressed as ng/2x104 cells.
Measurement of total lactic acid content
Lactic acid concentration in the lysed cells and exposure media was measured by commercial diagnostic kit (Randox, (UK). In short, lactate in the presence of oxygen is converted to pyruvate and hydrogen peroxide. The reaction is catalyzed by lactate oxidase. The hydrogen peroxide in the presence of peroxidase reacts with 4-amino antipyrine and n-ethyl-N-(2-hydroxy-3-sulphopropyl)m-toluidine to yield a purple colored product that was measured spectrophotometrically at 550 nm. The concentration of lactic acid was calculated using a standard curve and expressed as μM/2x106 cells.
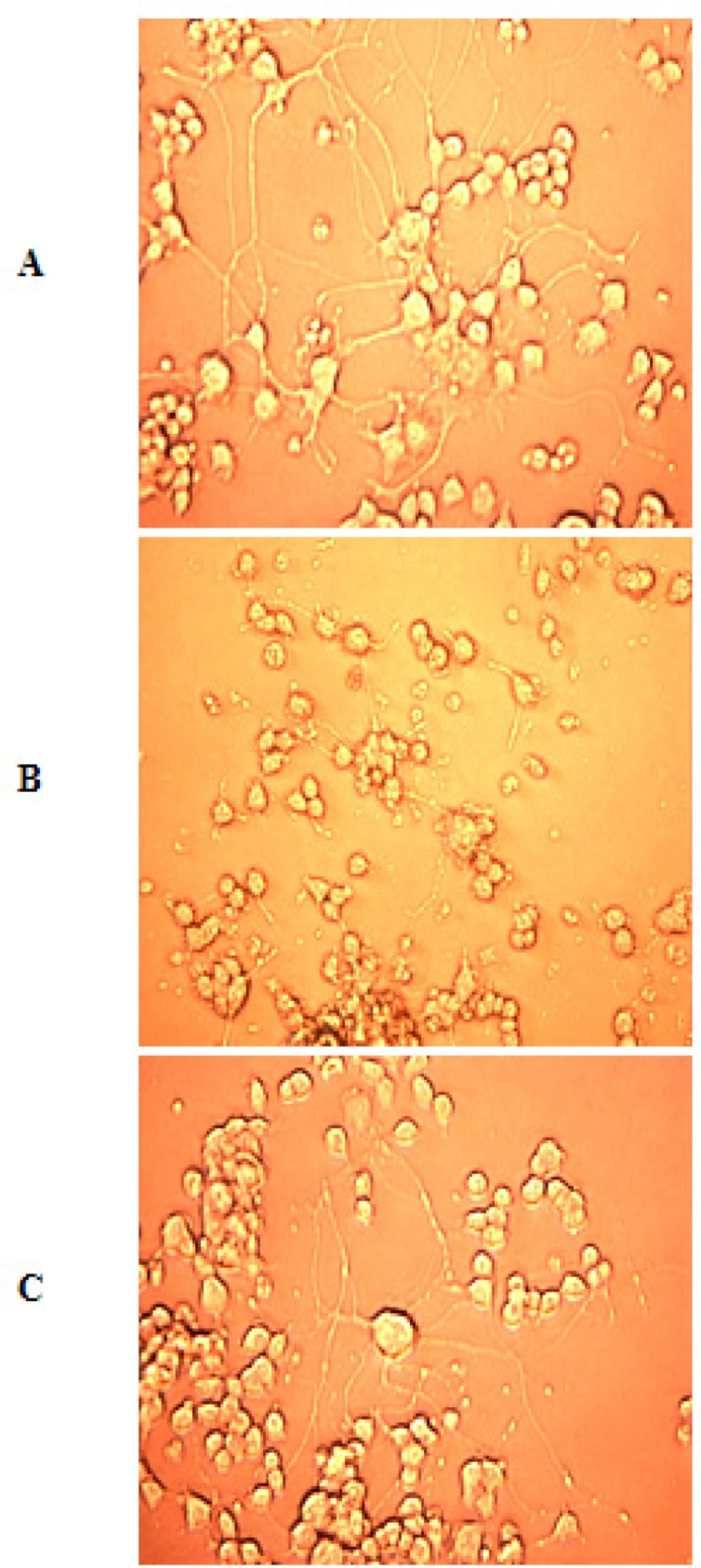
Photomicrographs
After ischemia-reperfusion photomicrographs were taken with the help of Inverted microscope (Leica DMIRB) to find out the morphological changes after ischemia.
Statistical analysis
For all antioxidant assays, all data are shown as mean ± SEM from three extraction replicates, each run in duplicate. Correlation and regression analysis of antioxidant activity and total phenolic content was carried out using SigmaPlot 2000 Software. For in-vitro methods, each experiment consisted of three separate plates from the same culture which were averaged together (n = 1). Each experiment was repeated three times using different cultures and the results expressed as mean ± SE of three different experiments. The statistical analysis was performed using one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls multiple comparison test. Statistical significance was drawn at p < 0.05.