Materials
Mefenamic acid was obtained from Smart (Smart Pharmaceutical Company, Ningbo China), ethyl cellulose 48 cP and cellulose acetate phthalate were obtained from Sigma-Aldrich (Sigma-Aldrich, USA) and chloroform, cyclohexane, tris buffer (pH of 9), Span 80, Liquid paraffin, n- hexane, acetone, ethanol, orthophosphoric acid, sodium lauryl sulfate and sodium hydroxide were obtained from Merck (Merck, Germany). All solvents and reagents were of analytical grade.
Method
Preparation of MA microparticles with CAP and/or EC polymers
Microspheres were prepared through oil-in-oil (O1/O2 emulsion solvent evaporation method) using different ratios of MA to CAP and/or EC ratios (as shown in
Tables 1 and
2). Liquid paraffin is preferred as an appropriate dispersing medium to ethyl alcohol and acetone, because when a solvent with a dielectric constant about 10 or above is used, non-polar liquid paraffin is preferred (
24,
25). Acetone is a unique organic solvent which is polar, water-miscible and oil-immiscible. All other organic solvents like methanol, ethyl alcohol, ethyl acetate, acetone, dimethyl sulfoxide and tetrahydrofuran are oil-miscible and do not form emulsions of the polymer solution in oil (
6,
9). MA was dispersed in 10 mL of the mixed solvent system consisting of acetone and ethyl alcohol in a 9 : 1 ratio (polymer solvent). The drug suspension was then emulsified in a liquid paraffin/span 80 solution under stirring at 600 rpm (Model RZR-2000; Heidolph Elektro, Kelheim, Germany) for 20 min. Then, 50 mL of chloroform or cyclohexane (non-solvent, for CAP and/or EC, respectively) was added to harden the microspheres and stirring was continued for a further 20 min. Next, the hardened microspheres were collected by filtration and washed with three portions of 30 mL of non-solvent to remove any remained oily phase, and then was air dried for 12 h.
Determination of loading efficiency and production yield (%)
Drug amount in microspheres was determined by dissolving 20 mg of each sample in 10 mL acetone while stirring using a mechanical stirrer at 500 rpm for 30 min. The drug concentration was determined spectrophotometrically (UV-160, Shimadzu, Japan) at 285 nm. All experiments were done in triplicate.
The loading efficiency (%) was calculated according to the following equation:
Loading efficiency (%) = (actual drug content in microparticles/theoretical drug content) × 100
The production yield of the microparticles was determined through accurately calculating the initial weight of the raw materials and the last weight of the polymeric particles obtained. All of the experiments were performed in triplicate.
Particle size analysis
A laser light scattering particle size analyzer (SALD-2101, Shimadzu, Japan) was used to determine the particle size of the drug and microparticle formulations. Samples were suspended in distilled water contained in a 1 cm cuvette and stirred continuously during the particle size analysis. Each sample was measured in triplicate.
Scanning electron microscopy
Surface morphology of microparticles was observed with a scanning electron microscope (LEO 440i, England) operating at 15 kV. The samples were mounted on a metal stub with a double adhesive tape and coated under vacuum with a platinum/palladium alloy using metallizer.
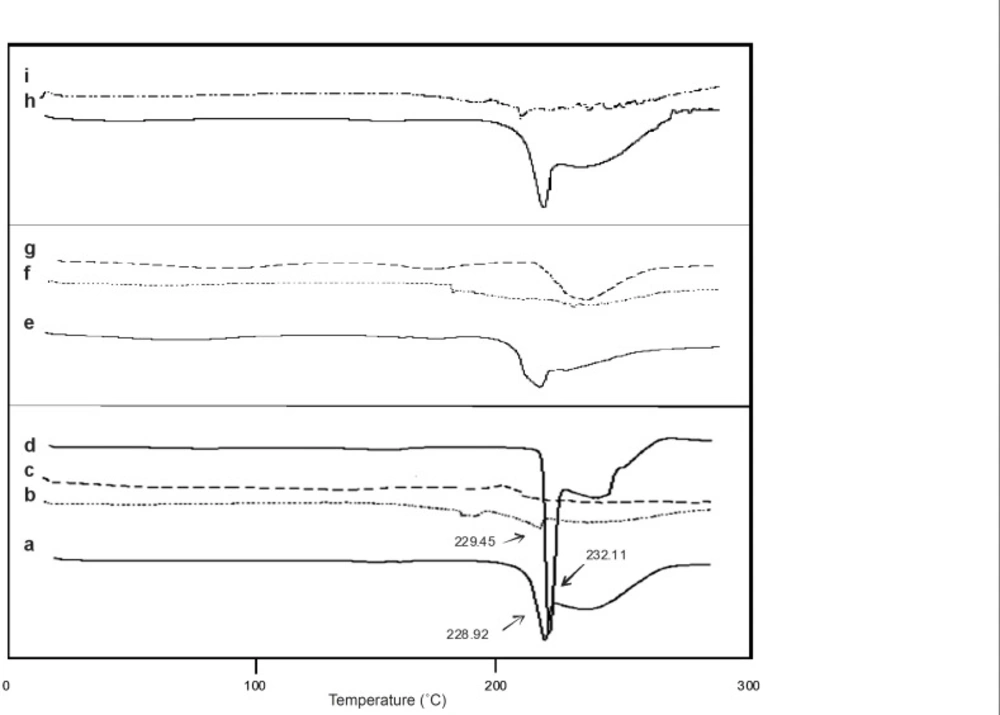
Differential scanning calorimetry (DSC)
DSC analysis (thermograph) of samples was done using DSC 60 instruments (Shimadzu, Japan). The samples were weighed into aluminum pans, which were closed with a pin-holed lid. Thermograms were recorded under nitrogen atmosphere from ambient to 300°C at a heating rate of 10˚C per min.
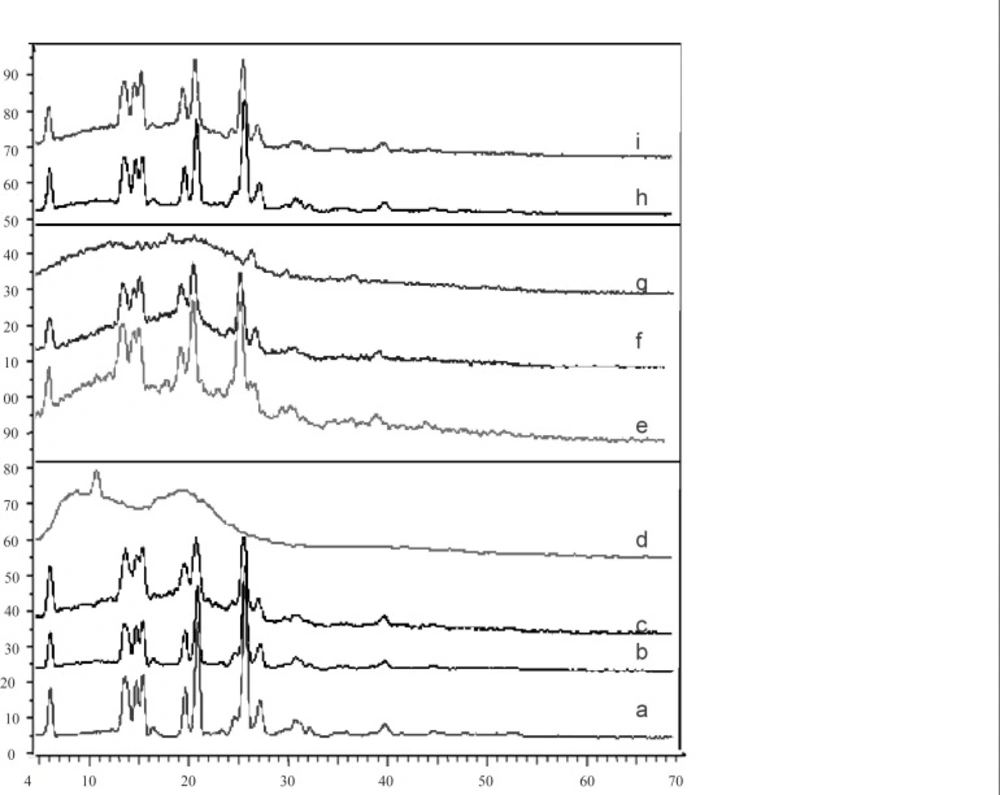
Powder X-ray diffractometry (X-RPD)
X-ray diffraction analysis was performed (Siemens D5000, Munich, Germany) using a nickel-filtered CuKα radiation (a voltage of 40 KV and a current of 20 mA). The scanning rate was 2°/min over a 2θ range of 20-60° and with an interval of 0.02°.
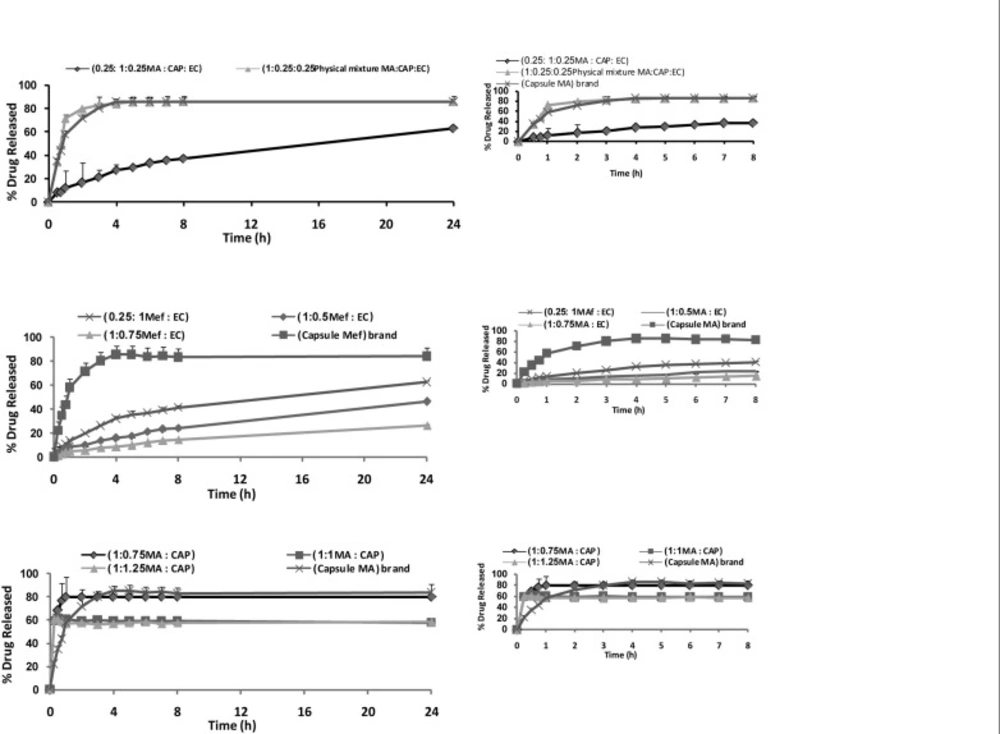
Dissolution studies
Dissolution was carried out using a USP basket method at 37°C and 100 rpm, in 900 mL of Tris buffer (pH 9). Microspheres (containing CAP and EC polymers separately and mixture with 1 : 1 ratio, respectively) were placed in the apparatus. Four mL of suspension was withdrawn at appropriate intervals (0. 5, 0.75, 1, 2, 3, 4, 5, 6, 7, 8 and 24 h) and each sample was returned into the apparatus. The samples were filtered through the 0.45 μm filters and used for the spectroscopic determination of the drug. Drug concentration in the samples was measured by UV spectrophotometric analysis at 285 nm. Each experiment was repeated three times.