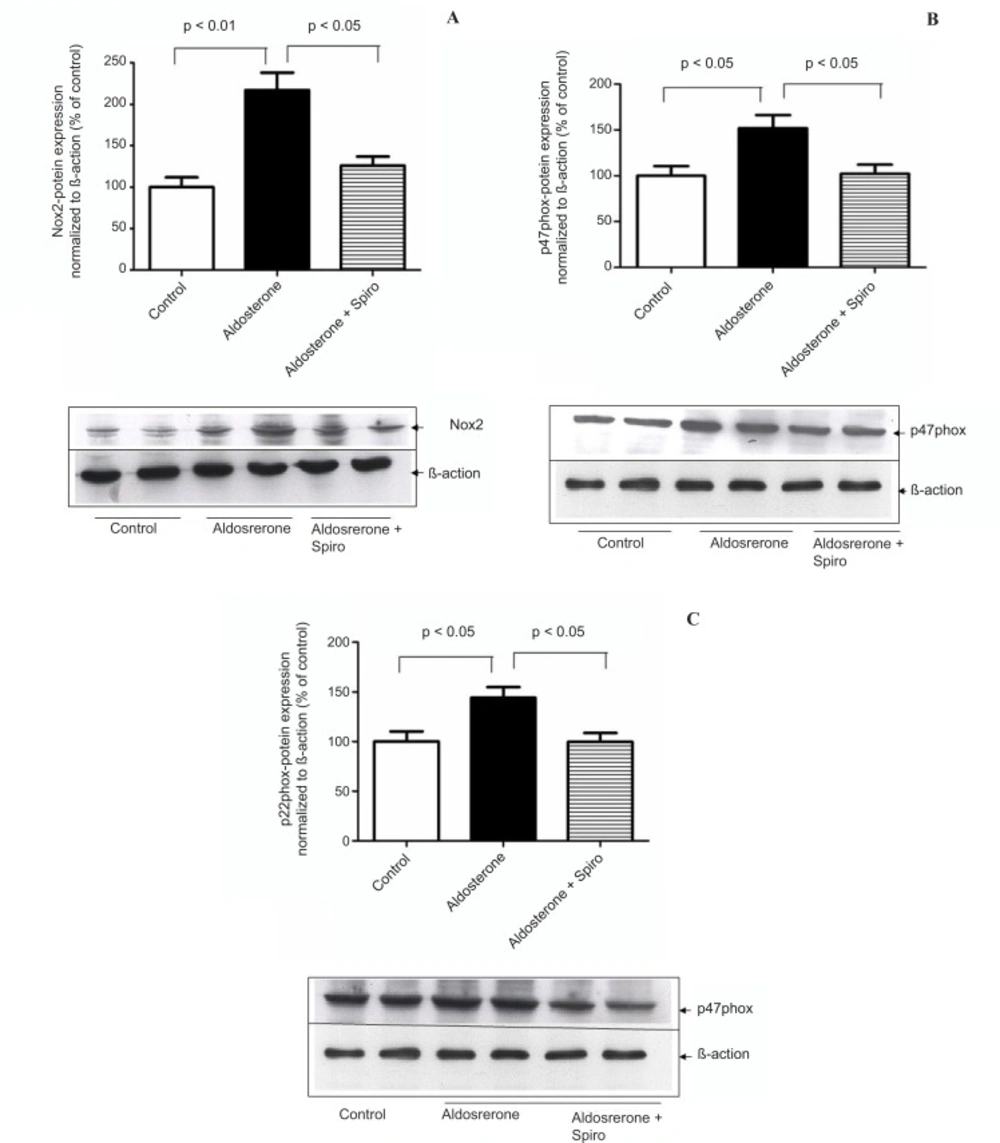
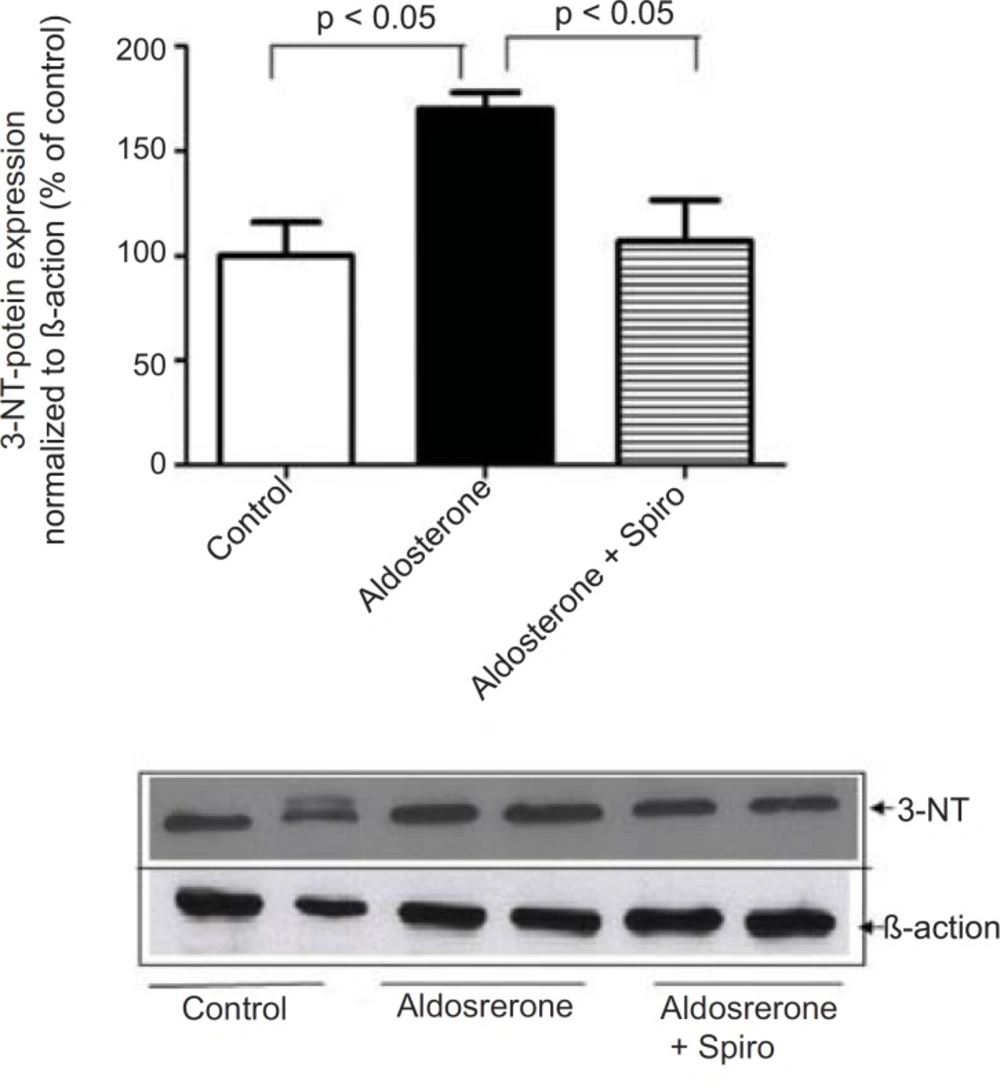
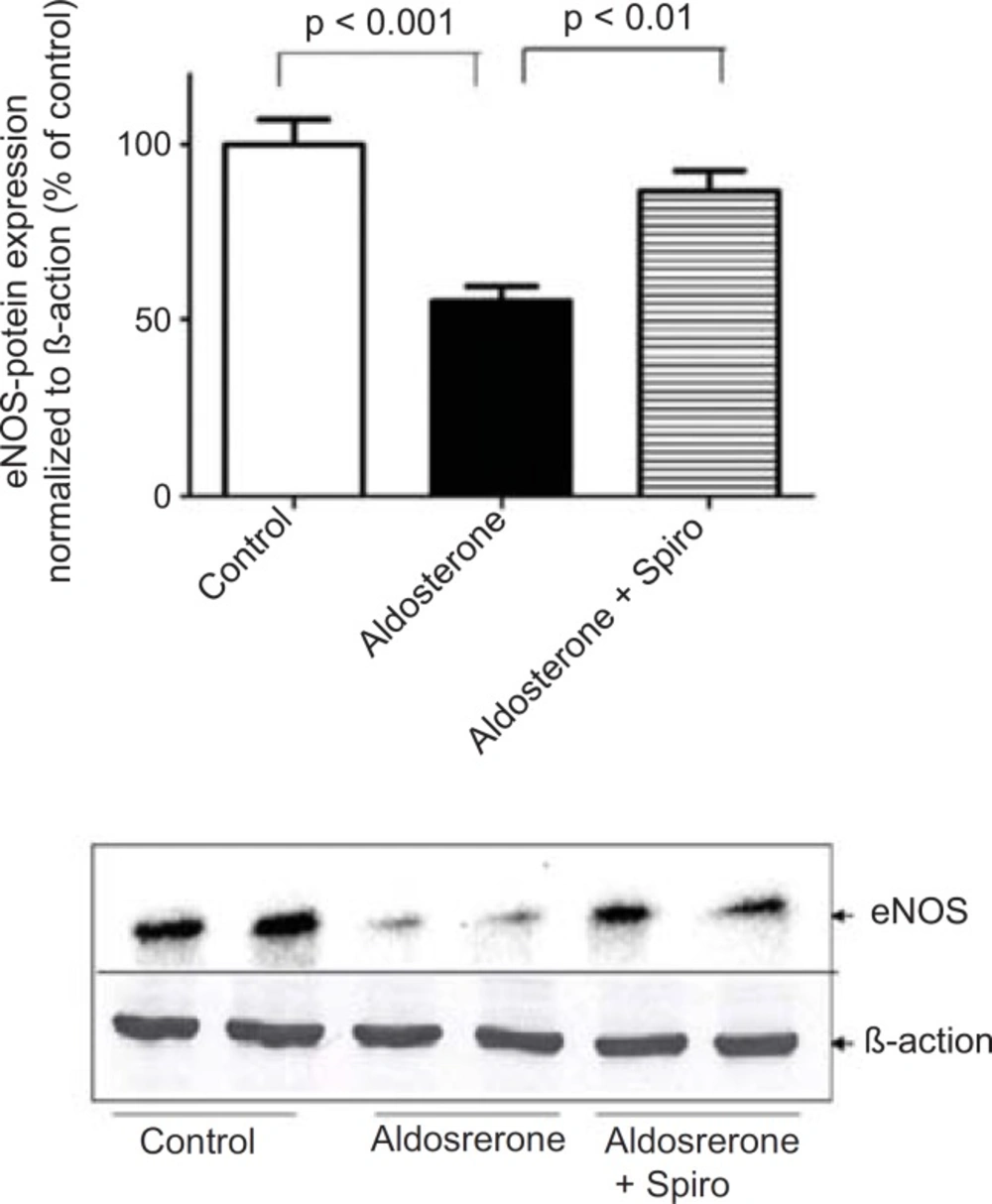
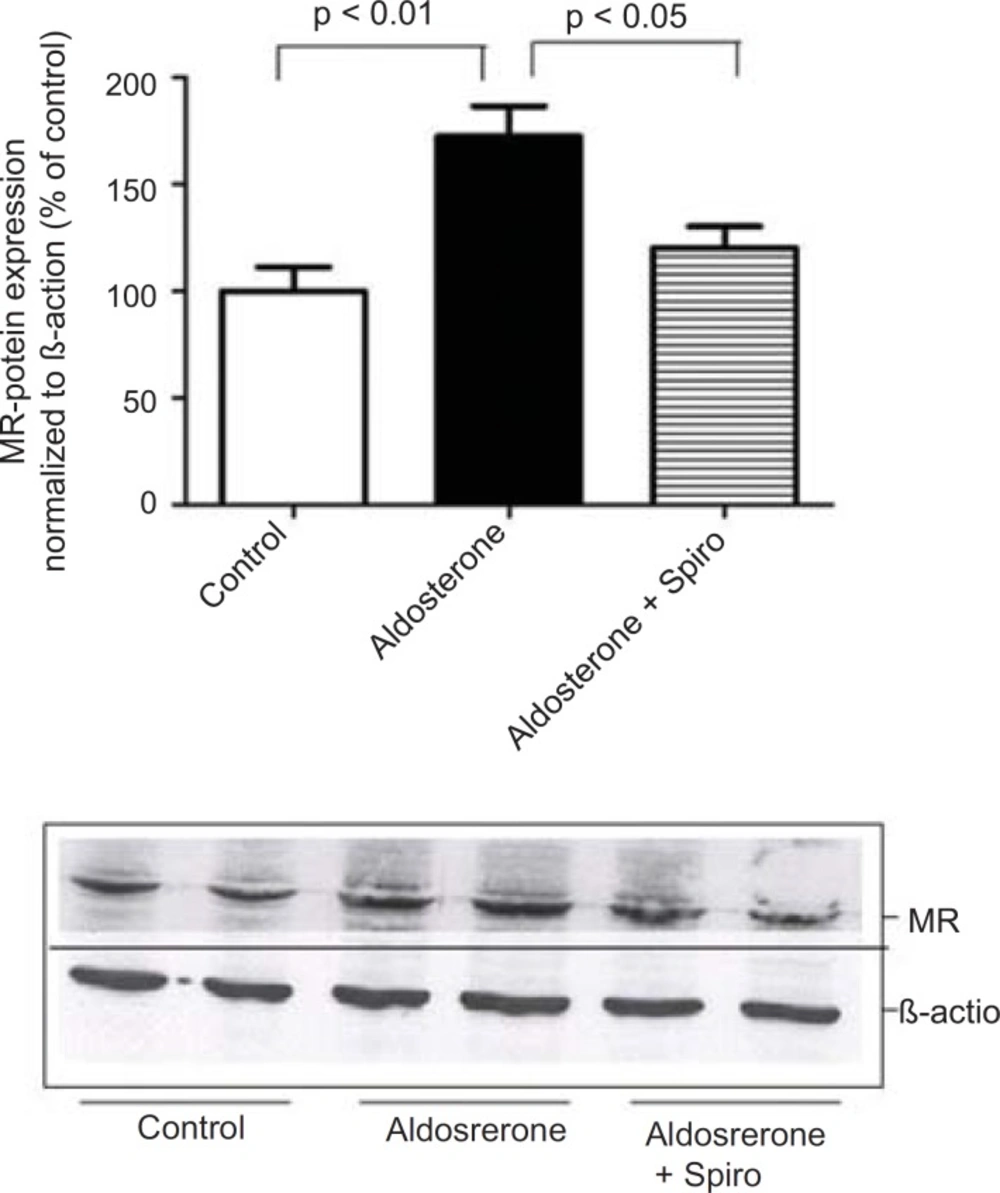
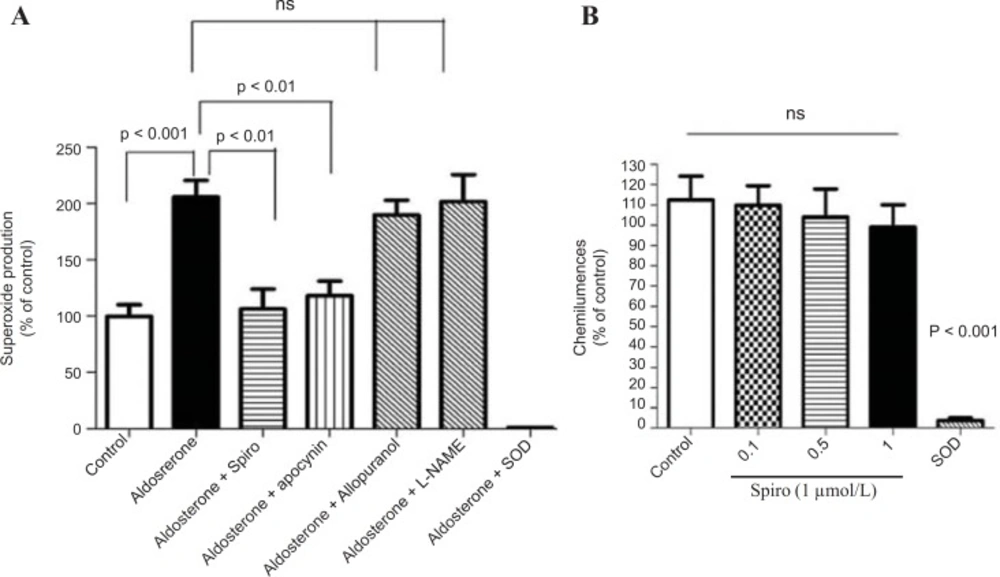
The present study showed that stimulation of the HUAECs with aldosterone resulted in a significant elevation of NADPH oxidase expression and activity. These changes were inhibited by the MR antagonist, spiro. We used a 100 nmol/L dose of aldosterone which is a close approximation to the
in vivo situation, particularly under hyperaldosteronism conditions. A growing body of evidence indicates that ROS are implicated in many pathophysiological processes including scavenging of endothelium-derived nitric oxide (NO) (
23), and prevention of its protective signaling functions (
24,
25). Although ROS may derive from mitochondria, xanthine oxidase, cyclooxygenase, uncoupled NO synthase, heme oxygenases, or peroxidases, it has been frequently shown that NADPH oxidasesare the primary producers of ROS in vascular tissues (
26,
27).Recent evidence suggests that NADPH oxidases, the only known enzyme family solely dedicated to ROS production, may be a key player. In harmony with this concept, we could not detect any significant role of xanthine oxidase and uncoupled eNOS in total ROS production. As the respective inhibitors, allopurinol and L-NAME, did not affect ROS signals in HUAECs stimulated by aldosterone. In contrast, the NADPH oxidase inhibitor, apocynin reduced ROS formation supporting the hypothesis that NADPH oxidases are indeed a major source of vascular oxidative stress in our system model.
Previous studies have suggested that ROS produced by NADPH oxidase mediate many angiotensin II effects in the cardiovascular system (
14,
26). Several reports suggest an important role for aldosterone in the regulation of NADPH oxidase. Recently, it has been reported that aldosterone increases NADPH oxidase expression in the vasculature (
28). Systemic administration of aldosterone increases oxidative stress in the heart, vasculature, kidney and increases macrophage NADPH oxidase (
29). In addition, MR activation contributes to angiotensin II-mediated activation of NADPH oxidase in the heart and aorta (
30). Furthermore, exogenous aldosterone stimulates aortic expression of p22
phox and Nox2 through an MR-dependent mechanism and of p47
phox mRNA through both an angiotensin type 1 receptor and MR-dependent mechanisms (
31). In this regard, the present study demonstrates a marked upregulation of Nox2, in contrast to Nox4 that did not exhibit any change. Accordingly, Nox2 might be considered as the relevant isoform involved in aldosterone-mediated NADPH oxidase activation. On the other hand, the cytosolic component of the p47
phox component was shown to havea pivotal role in the regulation of enzymatic activity. It has been reported that the hypertensive response and productionof vascular superoxide was markedly blunted in p47
phox knockoutmice (
32). It has been also shown that aldosterone induced NADPH oxidase activation and membranous translocation of p47
phox in HUAECs (
33). In this context, the current study shows that aldosterone increased thep47
phox protein level. Thus, aldosterone can increase ROS production in HUAECs by activating NADPH oxidase viap47
phox translocational regulation.
NADPH oxidase catalyzes the one-electron reduction of molecular oxygen to a superoxide anion which can react with nitric oxide to form short-lived peroxynitrite. Peroxynitrite forms stable 3-NT-conjugated protein moieties (
24). In this context, aldosterone was found to increase in the 3NT content as a biochemical marker of oxidative stress and this was significantly inhibited with pretreatment using spiro, supporting a potential role of MR in oxidative stress. Spiro was able to inhibit aldosterone-mediated NADPH oxidase activation, suggesting MR might be considered as upstream of NADPH oxidase. Specificity was demonstrated by means of the xanthine/xanthine oxidase assay, where antioxidative effects and flavoenzyme inhibition were excluded. Based on these finding, the present study demonstrates that spiro can antagonize aldosterone-mediated superoxide production and this is attributable to its inhibition of the NADPH oxidase enzyme. It has been reported that superoxide production reduces nitric oxide bioactivity while reducing the expression of nitric oxide synthase (
34). Evidence of endothelial dysfunction was seen in isolated renal artery segments and aortic rings from rats exposed to a model of excessive MR stimulation. In animals treated with spiro, normal endothelial function was restored (
8,
35). Similarly, in rabbits fed a proatherosclerotic diet, treatment with spiro normalized superoxide formation and improved endothelial function (
36). In healthy male volunteers, aldosterone has been shown to cause acute endothelial dysfunction (
37).
Notably, spiro significantly antagonized the inhibitory effect of aldosterone on eNOS expression. Importantly, using MR-reactive antibodies and western blotting we investigated MR protein expression in the HUAECs in response to aldosterone. This suggests that MR was the main receptor mediating the pro-oxidative effect of aldosterone in the current investigations. The present findings are in agreement with the previous
in-vivo study demonstratingthat eplerenone (a specific MR antagonist) administration to hypercholesterolemic rabbits normalized superoxide generation, decreased NADPH oxidase activity to basal levels and nearly normalized endothelium-dependent vasorelaxation.(
10). A recent study showed similar inhibition of atherosclerosis when eplerenone reduced markers of oxidative stress including the ability of macrophages to oxidize low density lipoprotein, macrophage superoxide anion release and the susceptibility of low density lipoprotein to oxidation (
38).
Regarding the mechanism of action aldosterone, it has been reported that aldosterone binds intracellular MR and translocates it to the nucleus, where it binds to its ligand and interacts with the regulatory region of target gene promoters (
39). In contrast, aldosterone might have a non-genomic effect within minutes (
40). Importantly, we could not detect ROS production within 2 h of stimulation by aldosterone, it took much longer as was previously reported (
28,
35). However, some reports showed that aldosterone induces ROS production through the activation of NADPH oxidase within 30 min (
41,
42). Such a discrepancy might be attributed to using different cell types or different experimental conditions.
In conclusion, the present results demonstrate that aldosterone stimulates NADPH oxidase-mediated oxidative stress thereby reducing eNOS expression. In addition, the MR antagonist, spiro appears to play a potential role in inhibiting these consequences. Thus, the current study suggests that NADPH oxidase might act as key regulator in aldosterone--mediated oxidative stress, thereby reducing eNOS expression. This study adds a new dimension to the understanding of the role of aldosterone in the activation of NADPH oxidase in human endothelial cells and supports the notion that aldosterone can induce the dysregulation of endothelial cells. This knowledge may lead to novel strategies for the prevention of oxidative stress and endothelial dysfunction via the blocking of MR.