Materials
Poly (D, L-lactic-co-glycolic acid) (PLGA; lactic–glycolic acid ratio; 50:50) in a molecular mass range of 54-69 kDa (RG 505) were purchased from Sigma (Germany). Polyvinyl alcohol (PVA) with 98-99% degree of saponification, 25-30 mpas viscosity, the density of 1.25 g/cm3, and MW of 74.8 kDa were obtained from NipponGohsei (Japan). The recombinant IFN-λ1 (100 ng/mL) was purchased from Research and Development systems (USA). Quant-iT ™ PicoGreen dsDNA assay kit was purchased from Invitrogen (USA). Endofree plasmid Giga Kit was purchased from Qiagen (Germany). Human Embryonic Kidney cell line (HEK293T) were obtained from Pasteur Institute (Iran). Mouse macrophage cell line (RAW264.7) was obtained from Faculty of Veterinary Medicine of Tehran University (Iran). Cervix carcinoma cell line (Hep2-C) and Encephalomyocarditis virus (EMCV) were obtained from Food and Drug Administration (Iran). The expression vector of pcDH-GFP(pGFP) encoding green fluorescent protein was kindly gifted by Prof. H. Soleimanjahi. All other chemicals and solvents were common laboratory-grade reagents.
Methods
Cloning and expression of pIFN-λ1
Cloning and expression of pcDNA3.1-IFN-λ1(pIFN-λ1) encoding interferon lambda-1 were done in our previous study (
26). In summary, total RNA from human monocyte-derived dendritic cells stimulated with 100 ng/mL of LPS was extracted and cDNA synthesized. PCR was performed by specific primers of IFN-λ1 mRNA and cloned inside the PTZ57R /T vector and then subcloned into pcDNA3.1 + using KpnI and BamHI restriction endonucleases. After confirming the IFN-λ1 cDNA sequence by Nucleotide BlAST at NCBI, pIFN-λ1 transfected into HEK293T by the polyfect reagent, the expression of IFN-λ1 was confirmed by sandwich ELISA method (R&D system, USA). After transformation of pIFN-λ1 into
E.coli (DH5a) using Cacl2 and cultivated in a high scale, purification of pIFN-λ1 was performed with Endofree plasmid Giga Kit according to the manufacturer's protocols. Similarly, pGFP purification was done.
Antiviral assay of IFN-λ1glycoprotein
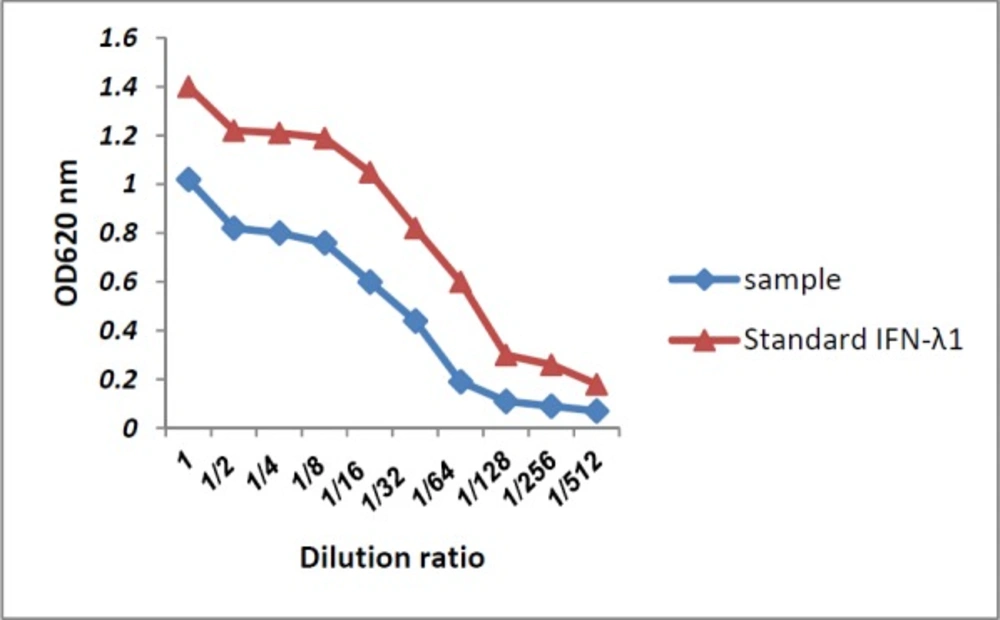
Before the encapsulation of pIFN-λ1 in PLGA, the biological activity of IFN- λ1 glycoprotein secreted in the supernatant of HEK293T cells transfected with the corresponding plasmid was analyzed by an antiviral assay as previously described (
27). In brief, Hep-2C cells were cultured in 100 µL of EMEM (Eagle's Minimum Essential Medium) supplemented with 10% fetal bovine serum (FBS) and 1% (v/v) pen/strep in 96-well plates at a concentration of 1.5 × 10
5 cells per well and incubated at 37 °C in 5% CO2 for 18 h. Two-fold serial dilution of IFN- λ1 secreted in the supernatant as well as recombinant IFN-λ1(100 ng/mL) were prepared using the culture medium and then transferred to the plate (Four wells were assigned to each dilution). After 24 h, the culture medium was aspirated and replaced by medium containing EMCV (3 × 10
7 PFU/mL) in all wells other than cells control. The plate was incubated at 37 °C and when 10% of cells of virus control were alive, staining process was started. At first, the plate was washed 3-4 times with PBS and stained with naphthalene black for 25 min at room temperature. After fixation with 4% formalin acetate for 10 min, cells were washed again and then 150 μL of 0.1 M NaOH was added and shaken for 30 sec. Finally, the absorbance was read at 620 nm by the spectrophotometer.
Preparation of pIFN-λ1-loaded PLGA NPs
The pIFN-λ1 was encapsulated in PLGA by the double-emulsion-solvent evaporation technique. For this purpose, 300 μg of pIFN-λ1 that was dissolved in 200 μL of Tris-EDTA buffer with pH 8.2 was added to 30 mg of PLGA which was dissolved in 1000 μL of chloroform and emulsified using microtip probe sonicator for 30 sec on the ice at 30% amplitude. This primary-water-in-oil emulsion was added dropwise to 6 mL of 1% (W/V) aqueous solution of PVA and sonicated for the 60s at 70% amplitude to form secondary-water-in-oil-in-water emulsion. In order to evaporate the chloroform, the secondary emulsion was agitated using a magnetic stirrer for ~18 h at 150 rpm. Then it was centrifuged for 10 min at 17000×g. The first supernatant was collected to calculate the non-encapsulated plasmids in the NPs. In order to remove PVA, pIFN-λ1-loaded NPs (F1 formulation) were washed twice with double-distilled water and resuspended in double distilled water. Finally, NPs were lyophilized for 24 h and stored at -70 °C for further investigation. Simultaneously, the same method was also used to produce empty NPs (F2 formulation) and the pGFP-loaded NPs. It is necessary to mention 300 μg of pGFP was also used.
Encapsulation efficiency, loading capacity, and production yield estimation
The standard calibration curve of DNA concentration versus fluorescence intensity was drawn according to Quant-iT ™ PicoGreen® dsDNA assay kit, with and without of PVA, using a fluorimeter (Biotek, Senergr HT, Winooski, VT, USA) at wavelengths of 485/520 nm. The first supernatant after centrifuge was collected and diluted in TE buffer and the non-encapsulated pIFN-λ1s content was quantified using the calibration curve. This content was subtracted from the total amount of pIFN-λ1 used in the encapsulation process (initial pIFN-λ1) and was considered as encapsulated pIFN-λ1s.The principles of calculating all three concepts (Encapsulation efficiency, loading capacity, and production yield) were taken according to the equations used by Khalid Mohamed El-Say (
28).
Morphological analysis of the NPs
In order to evaluate the morphology of pIFNλ1-loaded NPs and empty NPs, 1 mg of the NPs were added to 1 mL of distilled water and sonicated for 60 sec, and the resulting suspension was deposited on a scanning electron microscopy stub as a drop. After good drying at RT, NPs were coated with gold particles and their morphology were determined by a scanning electron microscope (SEM) (EM3200, KYKY, China) at an accelerating voltage of 26 kV with 10.0 KX magnitude. Three months after the storage of the lyophilized pIFNλ1-loaded NPs at -70 °C, they were re-examined by a transmission electron microscope (TEM) (ZIESS, EM900, PHILPS, Germany). For this purpose, the suspension of pIFNλ1-loaded NPs was added to a carbon grid. After drying at RT, the images were provided.
Size, Size distribution and Zeta potential measurement of the NPs
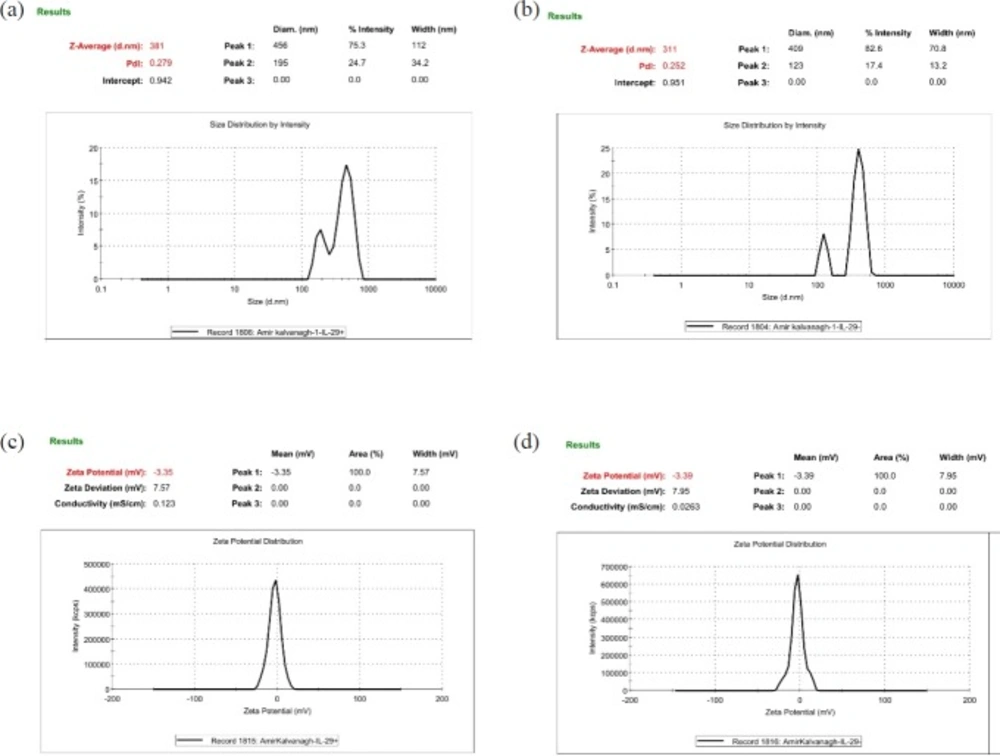
To evaluate the size, size distribution and, zeta potential properties of developed NPs, 0.7 mg of each lyophilized NPs were resuspended in sterilized distilled water using a bath sonicator (Ultrasonic Cleaner Set, WUC-D10H, Korea) for 3 min at 20% power. The zeta potential and size distribution of resuspended NPs were documented by dynamic light scattering method (Malvern Zetasizer ZS, UK). Size distribution and Zeta potential of the pIFNλ1-loaded and empty NPs were simultaneously determined.
In-vitro release pattern of pIFN-λ1 from the NPs
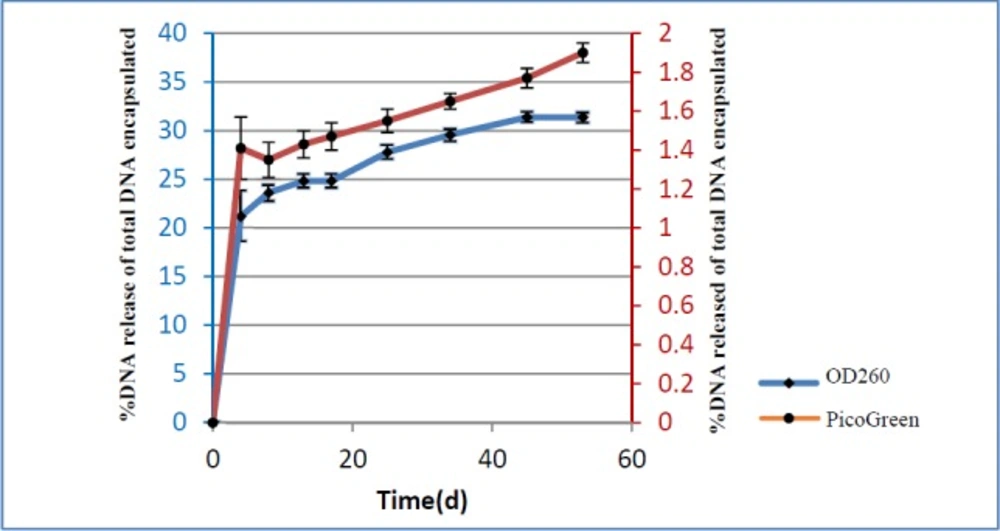
In order to find out the pattern of pIFN-λ1 release from the NPs, 1.5 mg of lyophilized pIFN-λ1-loaded NPs in 1 mL of PBS using bath sonicator were resuspended inside the DNase-RNase free tube at pH 7.2. The tubes were placed vertically in a shaker incubator (100 rpm) at 37 °C. At defined intervals (5 days in between), the samples were centrifuged for 5 min at 14000×g and supernatants were collected in new clean tubes. Immediately, the concentration of the released plasmids into the supernatant was calculated at 260 nm. The remaining supernatants were transferred to -20 °C and the NPs were resuspended in 1 mL of new PBS with pH 7.2 and placed back to a shaker incubator in the same condition. The concentration of all samples collected at -20 °C during 60 days was again measured with PicoGreen assay Kit. In fact, release pattern was investigated by two methods (spectrophotometry and fluorimetry). All release experiments were performed in triplicate.
| Formulation | Name | pIFN-λ1(µg) | Tris-EDTA buffer (μL) | PLGA(mg/mL) | PVA%(w/v) | Average ParticleSize (nm) ± SD | Average PolydispersityIndex ± SD | Average ZetaPotential (mV) ± SD |
|---|
| F1 | The pIFN-λ1-loaded NPs | 300 | 200 | 30 | 1% | 380 ± 3 | 0.26 ± 0.02 | 3.3 ± 7.6 |
| F2 | Empty NPs | 0 | 200 | 30 | 1% | 310 ± 4 | 0.24 ± 0.01 | 3.3 ± 7.9 |
| The concentration of empty NPs |
|---|
| Cell lines | 125 (μg/mL) | 250 (μg /mL) | 500 (μg /mL) |
| RAW264.7 | 97 ± 3% | 96 ± 3% | 95 ± 2% |
| HEK293T | 95 ± 2% | 94 ± 1% | 93 ± 1% |
| NPs | Encapsulation efficiency (%) | Loading capacity (%) | Production yield (%) |
|---|
| The pIFN-λ1-loaded NPs | 75 ± 5 | 0.83 ± 0.06 | 89 ± 0.5 |
The SEM images of NPs prepared using the double-emulsion-solvent evaporation technique. (a) The pIFN-λ1-loaded NPs, and (b) empty NPs. (c and d) The TEM images of the pIFN-λ1-loaded NPs prepared using the double-emulsion-solvent evaporation technique after 3 months of storage at -70 °C
Comparing of the pIFN-λ-1-loaded NPs and empty NPs in terms of size, PdI, and zeta potential. All of the parameters are obtained by Zeta Sizer. (a and c) The NPs formulated with F1 formulation and (b and d) the NPs formulated with F2 formulation
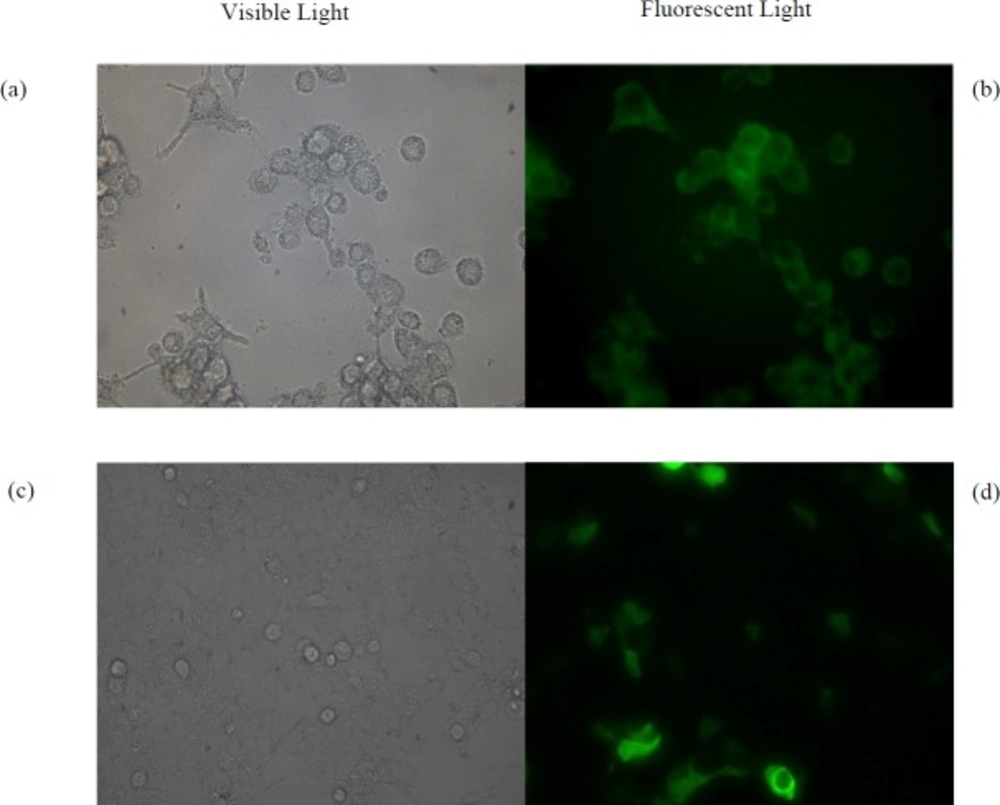
(a) Microscopic images of RAW264.7 cells under visible light and (b) fluorescent light treated by the pGFP- loaded NPs. (c) Microscopic images of HEK293T cells under visible light and (d) fluorescent light treated by free pGFP using calcium phosphate method
Release patterns of pIFN-λ1 from NPs were measured by Picogreen assay and UV absorbance at 260 nm
Antiviral activity of IFN-λ1/IL-29 secreted to the supernatant of HEK293T cells (sample) compared to recombinant IFN-λ1 using the CPER test
Cellular uptake and bioactivity of the NPs
The HEK293T and RAW264.7 Cells were chosen as non-phagocytic and phagocytic cell lines, respectively. Both of them were grown in 2 mL of DMEM Medium supplemented with 5% fetal bovine serum (FBS) and 1% (v/v) pen/strep in 12-well plates at a concentration of 1.25 × 105 cells per well for 22 h. Then the medium was aspirated, the cells were washed and 1 mL of fresh medium was added. After 2 h, cells were treated with 0.5 mg of suspended pGFP-loaded NPs by which the same method described for pIFN-λ1-loaded NPs were prepared. After18 h treatment, in order to remove NPs that were not uptaken by cells, 80% of the culture medium was gently collected and replaced with fresh medium. Expression of GFP was examined under fluorescence microscope four days after treatment of cells with NPs. As a positive control, pGFP transfected by calcium phosphate method in both cell lines and after two days examined under a fluorescence microscope. Untreated cells were used as negative control. In addition to these controls, both of cell lines were treated with three different concentration of empty NPs (125, 250 and 500 µg/mL) to evaluate the toxicity of NPs fabricated by PLGA.