The accumulation of pro-inflammatory compounds released in glomerulonephritis could lead to inflammation. Inflammation as well as other factors including hypertension, hyperlipidemia, endothelial dysfunction, and proteinuria are associated with a higher risk for cardiovascular diseases (CVD) in glomerulonephritic patients (
9,
10). Based on published data, synthesis and secretion of pro-inflammatory cytokines have been increased during dialysis as a result of the interaction of PBMCs with dialytic membranes. So, dialysis is associated with increased severity of inflammation in glomerulonephritic patients undergoing hemodialysis (
11,
12). Multiple mechanisms, including backfiltration of contaminated dialysate to the blood compartment and increased expression of complement fragments, contribute to the secretion of cytokines during hemodialysis (
13,
14). Here, for the first time, the activation of NLRP3 inflammasome was considered as another mechanism involved in pro-inflammatory compounds release in glomerulonephritic patients undergoing hemodialysis. Although the formation and activation of NLRP3 were investigated in primary glomerulonephritis by Xiong
et al., no report has reported its involvement in chronic inflammation associated with dialysis (
8). In line with results obtained here, Granata
et al. observed elevated expression levels of NLRP3 inflammasome components in PBMCs derived from CKD patients undergoing dialysis treatment compared with those derived from healthy subjects (
7). Up to now, most investigations on the role of NLRP3 inflammasome in kidney diseases have been conducted either in animal models or in cultured cells, however, the data collected from humans are infrequent. For example, in the mice model, NLRP3 was shown to be activated in UUO-induced renal impairment. Compared with wild-type mice, their results revealed the reduced caspase-1 activation and IL-1β/IL-18 maturation in Nlrp3
-/- mice (
15).
Here, we also observed a statistically significant increase in IL-1β serum level of glomerulonephritis-HD patients which can further support the activation of the NLRP3-ASC-caspase-1 axis in these patients. In line with this observation, Xiong et al reported that in PGN patients, the serum level of IL-1β was markedly increased and positively correlated with kidney damage (
8). Although based on
in-vitro studies, NLRP3 inflammasome may have a role in the pathogenesis of renal diseases via overexpression of proinflammatory cytokines including IL-1β and IL-18, several pieces of evidence show that the NLRP3 effect may be independent of pro-inflammatory cytokine production. Shigeoka
et al. demonstrated that the absence of NLRP3, but not the downstream inflammasome targets, was able to preserve kidney tissue from injury. These results revealed the non-classical direct effect of NLRP3 on renal tubular epithelium which was independent of pro-inflammatory cytokine production mediated by inflammasome induction. Consequently, various mechanisms may be involved in the pathogenesis of renal diseases mediated by the NLRP3 inflammasome (
16).
Two steps are currently known as major upstream mechanisms for NLRP3 inflammasome activation. The Toll-like receptor (TLR)/ NF-κB signaling pathway is responsible to induce the first step and leads to overexpression of proinflammatory cytokines as well as NLRP3 inflammasome components which are relatively low in numerous types of cells. The second step leads to the assembly of a multi-protein complex including NLRP3, ASC, and pro-caspase-1. Reactive oxygen species (ROS) generation, lysosomal destabilization and rupture and the engagement of P2RX7 receptor by high concentrations of ATP are several mechanisms proposed for NLRP3 activation which leads to the activation of caspase-1 and the secretion of mature IL1β (
3,
17). In this study, no significant difference was observed in the mRNA expression level of the P2X7 receptor in PBMC isolated from glomerulonephritic patients compared with those extracted from controls. On the contrary, the P2X7 receptor was shown to be overexpressed in a rat model of glomerulonephritis and human lupus-related GN (
18,
19). Moreover, in an accelerated nephrotoxic GN mouse model, the expression level of P2X7 receptor protein was also increased (
20). Other molecular mechanisms such as ROS generation, lysosomal destabilization, and rupture may be involved here. NLRP3 inflammasome activation mediated by Damage-associated molecular patterns (DAMPs) such as extracellular matrix components and ROS in several renal diseases was previously reported. For example, in CKD-HD patients-derived PBMCs, Granata
et al. proposed that NLRP3 inflammasome activation could be possibly triggered by mitochondrial dysfunction which led to ROS production (
7). Additionally, NLRP3 inflammasome/caspase-1/mitochondria axis was reported to be able to mediate albumin-induced renal tubular injury. Although, based on results obtained here, P2X7 receptor may not be involved in inflammasome activation, , as previously shown, using selective P2X7 receptor agonists or antagonists as well as deletion of P2X7 receptor should be needed for researches investigated the exact role of P2X7 receptor in cellular mechanisms including NLRP3 inflammasome pathway activation. For example, the importance of the NLRP3 inflammasome pathway in the nephrotoxic nephritis model in the Wistar Kyoto (WKY) rat was firstly highlighted by Deplano
et al. Using P2X7 receptor-deficient model, they showed that P2RX7 activation could trigger the NLRP3 inflammasome pathway which was resulted in IL-1 and IL-18 release in macrophages (
21). However, the mechanism involved in NLRP3 inflammasome activation in the PBMC derived from glomerulonephritis-HD patients remains to be investigated.
Previously, the activation and production of IL-1β were shown to be related to the NLRP3 inflammasome activation in glomerulonephritic patients, however, the activation of other inflammasomes under glomerolonephritis has not been investigated before (
8). To understand the role of other inflammasomes in inflammatory status observed in glomerolonephritis-HD patients, we investigated the probable role of NLRC4 in the processing and activation of pro-IL-1β. We revealed that PBMCs isolated from glomerolonephritis-HD patients had elevated mRNA levels of NLRC4, suggesting the priming of NLRC4 inflammasome. In agreement, yuan et al reported an increase in NLRC4 expression in the kidney of DN patients (
4). Moreover, Guo et al found that
NLRC4 expression level was enhanced after kidney ischemia–reperfusion injury (IRI). They showed the role of T cell immunoglobulin domain and mucin domain‐containing molecule‐3 in NLRC4 inflammasome activation and regulation of the TLR‐4/ NFκB signaling pathway (
22). To elucidate the role of NLRC4 inflammasome in the glomerolonephritis development, the pro-IL-1β activation should be investigated after blockage of inflammasome/IL-1β signaling pathway. For example, in yuan et al study, NLRC4 deficiency in DN mice was shown to be associated with a decrease in the level of IL-1β expression in renal tissues (
4).
These results reveal, for the first time, an enhanced inflammatory status in glomerulonephritic patients undergoing hemodialysis treatment. We propose that the NLRP3-ASC-caspase-1 axis may have a role in IL-1β secretion and inflammation in glomerulonephritic patients undergoing hemodialysis treatment. Moreover, these data further show an increased expression of NLRC4 inflammasome and its possible involvement in inflammation associated with hemodialysis in glomerulonephritis-HD patients. In summary, these findings provide new insights into molecular mechanisms underlying chronic inflammation in HD- glomerulonephritic patients. Additionally, the NLRP3 inflammasome pathway could be attractive as a potential therapeutic target to effectively minimize or avoid severe complications in glomerulonephritic patients undergoing hemodialysis treatment.
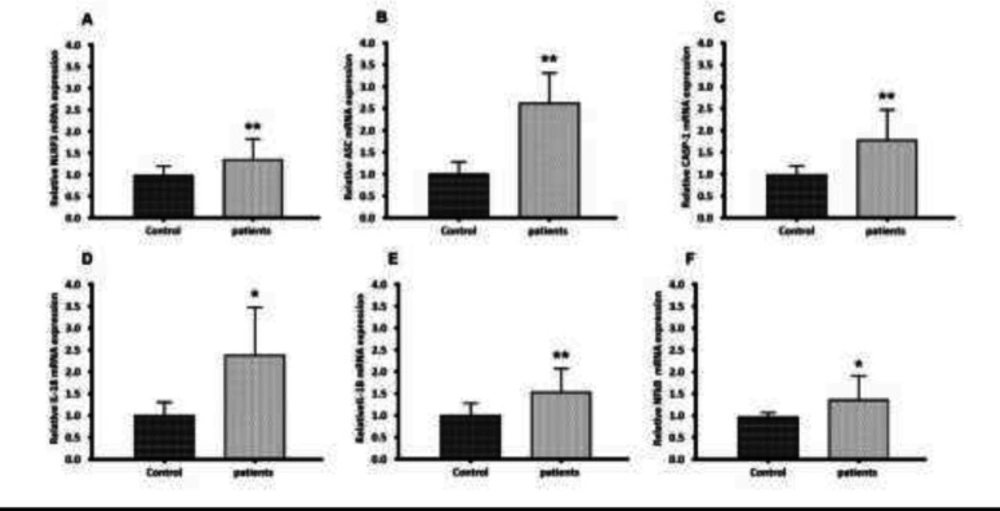
Gene expression of NLRP3, ASC, CASP-1, IL-18, IL-1β, and NFkB in PBMC from glomerulonephritis-HD patients compared to healthy controls. Histograms show the mRNA levels of (A) NLRP3, (B) ASC, (C) CASP-1, (D) IL-18, (E) IL-1β and (F) NFkB evaluated by RT-qPCR in PBMC derived from 28 glomerulonephritis-HD patients and 28 healthy controls. RT-qPCR results were normalized to GAPDH expression used as reference gene. For all genes, results showed higher expression levels in glomerulonephritis-HD patients compared to healthy controls (*p < 0.05, **p < 0.01)
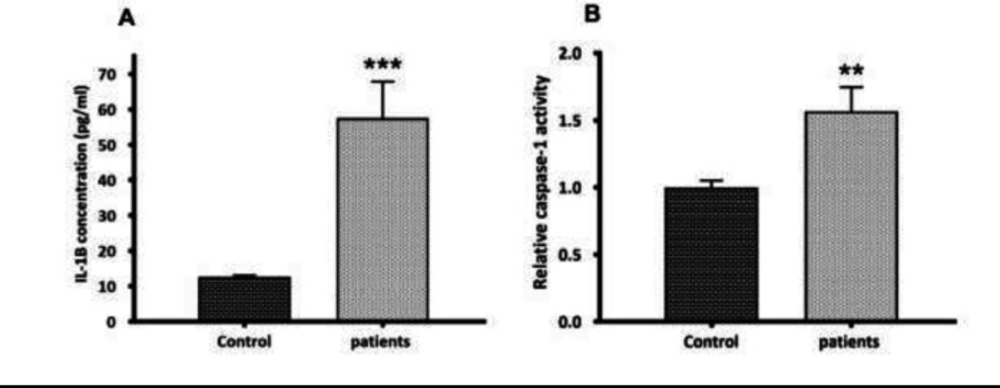
the second step in NLRP3 inflammasome activation. (A) Quantification of mature IL-1β secretion by ELISA. Compared to controls, an elevated serum level of IL-1β (4.58--fold increase) (p < 0.001) was observed in glomerulonephritis-HD patients. (B) Caspase-1 activity assay. The activity of caspase-1 was higher in glomerulonephritis-HD patients compared to controls (1.57-fold increase) (n = 10) (p < 0.01). Data represent mean ± SD
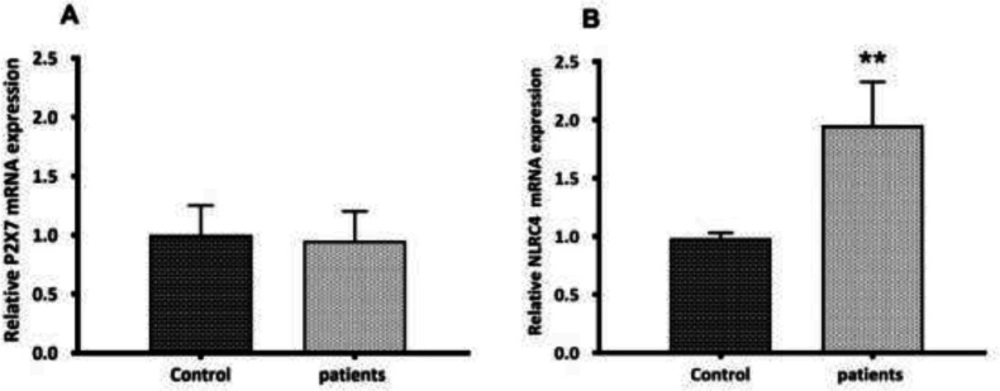
P2X7 and NLRC4 expression levels between PBMCs derived from glomerulonephritis-HD patients and those isolated from controls. Histograms show the mRNA levels of (A) P2X7, and (B) NLRC4 evaluated by RT-qPCR in PBMC derived from 28 glomerulonephritis-HD patients and 28 healthy controls. RT-qPCR results were normalized to GAPDH expression used as reference gene. No significant difference was observed in P2X7 expression level between two groups. For NLRC4 gene, results showed higher expression level in glomerulonephritis-HD patients compared to healthy controls (**p < 0.01).
| Genes | Size of PCR product (bp) | Primers (5'–3') |
|---|
| NLRP3 | 235 | Forward: CAGCAGATGGAGAGTGGCAAG Reverse: AAGCAGACACATCCGCCTTCT |
| NLRC4 | 140 | Forward: CACTTGTCTGACATTGGAGAGGGReverse: TTGTGAAGATTCTGAGCTAGGATTTTC |
| ASC | 100 | Forward: GGAAGGTCCTGACGGATGAGReverse: CAGTTCCAGGCTGGTGTGAA |
| CASP-1 | 139 | Forward: CAAGAATATGCCTGTTCCTGTGATReverse: GTCCTGGGAAGAGGTAGAAACATC |
| IL-1β | 129 | Forward: AGTGGCAATGAGGATGACTTGTTReverse: GCTGTAGTGGTGGTCGGAGATT |
| IL-18 | 160 | Forward: TGACCAAGTTCTCTTCATTGACCAReverse: CTCACACTTCACAGAGATAGTTACAGCC |
| NFkB | 118 | Forward: GCTACACAGGACCAGGGACAGTReverse: AGCTCAGCCTCATAGAAGCCATC |
| P2X7 | 246 | Forward: GCATCACCACCTCAGAGCTGTReverse: ACTGCCCTTCACTCTTCGGAA |