1. Background
The struggle against cancer continues to pose a global challenge across the world. Even though the standards of health care and rehabilitation and cancer survival rates have improved, liver cancer is the seventh most common cancer and the third leading cause of cancer-related death (1). It has an annual incidence of more than 800,000 cases and accounts for approximately 700,000 deaths each year (2). Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer, which accounts for more than 80% of all liver cancers (3). The burden and global age distribution of HCC vary greatly by gender, etiology, and geographic region because of differences in risk factor exposure (4). Viral Hepatitis is the predominant cause of HCC worldwide. Approximately, more than 75% of all cases of HCC are due to Hepatitis B and C Virus infections (5). The regions that have a higher burden of viral hepatitis have a higher load of HCC (6). Therefore, the significant increase in incidence and death rates of HCC is highly attributed to the increase in infections from HBV and HCV. Other factors that highly increase the risk of liver cancer development include the use of tobacco (smoking), heavy consumption of Alcohol, and obesity (overweight) (7).
The human genome is composed of coding and non-coding regions. It has been found that only a small fraction (about 1%) of human DNA is protein-coding while the remaining large portion (about 99%) is non-coding DNA i.e. it does not code for protein (8). The non-coding regions of DNA contain regulatory elements. In cancers, there are genetic alterations (also known as mutations) in regulatory elements, which cause dysregulation of tumor suppressor genes called oncogenes (genes that protect the body from cancers). Somatic mutations that occur at a higher rate are called ‘driver mutations. Driver mutations can be present in genes that are involved in the maintenance of genome and chromosomal stability (9).
The analysis of non-coding regions is quite difficult. The challenges associated with the study of non-coding regions are unique and distinct from the challenges of the coding region. The driver mutations in non-coding regions play a significant role in the progression of cancer. Different approaches have been developed to identify candidate cancer driver genes but still, it is difficult to distinguish, which epigenetic and genetic changes are developing cancers. Many researchers investigated the role of regulatory mutations in non-coding regions and attempted to identify driver mutations in regulatory regions. In a study, recurrent non-coding mutations were identified within the TAL1 enhancer region in acute lymphoblastic leukemia. It suggested that there is an impact of mutations in the TAL1 enhancer region on the regulatory factors of disease (10). A similar study by Puente et al. discovered recurrent non-coding mutations in the enhancer region, which is close to the PAX5 gene in chronic lymphocytic leukemia (CLL) patients (11). Another important class of non-coding mutations includes mutations in functional RNA molecules (long non-coding RNA [lncRNA] and micro RNA [miRNA]). The lncRNA of MALAT1 was found to be mutated in breast cancer (12). The role of mutations in binding sites and non-coding DNA was identified by Katainen et al. In their study, frequent mutations were observed in CTCF/cohesion-binding sites in cancers. These results revealed that mutations at CTCF binding sites are significantly important in cancers (13). Some studies have identified recurrent somatic mutations in TERT promoter regions across various cancer patients. One study identified mutations in the TERT promoter region at known and novel sites, which suggested a significant role of regulatory mutations in diseases like cancers (14). The cancer types, in which mutations in TERT promoter regions were found to affect patient survival, include bladder cancer (15), gliomas (16), and renal cell carcinoma (17). Recently, a study by Schulze et al. identified TERT promoter mutations in alcohol-related hepatocellular carcinoma patients. In this study, these mutations were thought to be responsible for tumor progression (18). Some recurrent mutations in the promoter region of NFKBIE have also been identified in desmoplastic melanoma (19).
2. Objectives
The main objective of the present study is to identify novel genes that are not known to cause liver cancer. It also aims at identifying pathogenic alleles that may act as potential biomarkers for the prediction of liver cancer. It focuses on mutations that are reported in non-coding regions of human DNA. Bioinformatics tools are used to study genetic alterations associated with liver cancer at the molecular level. Liver cancer is mostly asymptomatic at early stages and symptoms usually begin to appear at later stages when a cure becomes difficult. Most patients fail to receive successful treatment because of late diagnosis of disease. So, for patients with no or few symptoms, there is a need for biomarkers that can detect liver cancer at early stages when treatment is possible. These biomarkers will also help reduce the risk of the development of liver cancer. The results of this study may help identify driver mutations and genes involved in liver cancer progression. The biomarkers (pathogenic alleles) identified in this study can be used in further studies for verification.
3. Methods
The data file (Cosmic non-coding variants) of genome version GRCh38 was downloaded from the catalogue of somatic mutations in cancer (COSMIC) database. The file contains complete data on non-coding mutations in different types of cancer. The first step was to filter out all the non-coding mutations that were reported in liver cancer. The complete methodology of this study is illustrated in Figure 1.

Figure 1.
Schematic diagram of the methodology followed for analysis of non-coding mutations. The arrows represent the final results. The lines represent files used for the corresponding operations. TF, transcription factors.
3.1. Identification of Consistent Mutations at HepG2 Transcription Factors Binding Sites
The consistent non-coding mutations were found, using customized Python code. The next step was to determine whether these recurrent non-coding mutations are at transcription factor binding sites (TFBS) or not. The data files of transcription factor binding sites for HepG2 cells were downloaded from UCSC ENCODE. The size of transcription factor binding sites that are obtained from ChIP-Seq experiments is large; therefore, to obtain significant results, this size was reduced to 100 base pairs. The TFBS files with actual and reduced sizes were, then, overlapped with consistent non-coding mutations individually to identify which transcription factor (TF) binds at consistent non-coding mutations. These mutations were, then, searched in the VISTA Enhancer Browser to determine whether they are part of identified human gene enhancer or not. The list of 1912 elements with enhancer activity for humans was downloaded from the Enhancer Vista browser. All downloaded files were of genome version GRCh37/hg19. These files were converted to genome version GRCh38/hg38, using UCSC Genome Assembly.
3.2. Significance of Reported Non-coding Mutations
The significance of all reported non-coding mutations was determined by calculating their scores and empirical P-value on the basis of consistency and the number of transcription factors that were binding. For scoring, equal points i.e., 5 were assigned to both. The highest consistency was found to be 410 and the minimum consistency was 1. Since the second highest consistency was 15, the mutation with consistency 410 was considered to be an outlier, and ranking was done from mutation with consistency 15. The maximum and minimum numbers of TF binding were 39 and 1. The following formula was used for scoring non-coding mutations
Where,
Maximum consistency = 15, Consistency score = 5, Maximum no. of TF binding = 39, TF binding score =5.
The statistical significance of the acquired results was determined by randomization. It was done to eliminate biases from the results. For this purpose, 10,000 random samples were selected from the complete file of non-coding variants. This file also had mutations with no TF binding in HepG2 cells. In this analysis, the cut-off value i.e., alpha for significance was set to be 0.05. The lower the P-value, the more significant the mutations are.
3.3. Association of Genes with Non-coding Mutations
The genes that were closer to a great number of non-coding mutations were identified. It was also analyzed whether these mutations were in the upstream region, downstream region, or within the coding region of these genes. The closest distance of mutation from the Transcription Start Site (TSS) of the corresponding gene was also found.
3.4. Mapping Non-coding Mutations to CTCF Binding Sites
CTCF is a transcription factor that acts as an activator, repressor, or insulator protein. It controls gene expression either by insulation of enhancers or by activating or repressing promoters as it can bind a wide range of sequences. This diversified role of CTCF led researchers to map its binding sites in different species (20). Therefore; mapping of non-coding mutations was done with HepG2 cells CTCF-binding sites. Before mapping, the clusters of non-coding mutations were made. For each cluster, the maximum distance between mutations was set to 100. It means the mutations that were within 100 base pairs were combined in one cluster. The overlapping clusters were also combined.
3.5. Graphical Analysis of Significant Non-coding Mutations and Clusters
The graphical profiles of important non-coding mutations and clusters were obtained from the UCSC Genome browser (https://genome.ucsc.edu). It provides annotations for the specific regions of a genome. This browser is highly customized and displays relevant information only. The regions showing variations in results were selected for analysis. Only a few HepG2 cells TF (CTCF, FOXA1, SP1, and SIN3A) were displayed from the regulation feature due to the limited window. The conservation track was also selected, which provided regions that were most likely conserved in different species.
3.6. Analysis of Ras/MAPK Signaling Pathway
The mitogen-activated protein kinase (MAPK) pathway plays a significant role in the survival and growth of cells. It regulates the expression of genes (21). It also regulates the replication of the hepatitis B virus. The replication of HBV is suppressed when this pathway is activated (22). Any abnormality in the Ras/MAPK signaling pathway may lead to resistance to apoptosis causing increased and uncontrolled cell proliferation. Different studies have shown its involvement in some cancers (23). Ras/MAPK is also activated in 50% to 100% of cases of primary liver cancer (HCC) (24). Therefore, it is considered a potential target for treating HCC. In this study, mutations reported near genes involved in the MAPK signaling pathway were identified.
4. Results
The Following Section Summarizes the Results Obtained from this Study.
4.1. Consistent Mutations at HepG2 Transcription Factor Binding Sites
The complete list of identified non-coding mutations is present in Appendix in Supplementary File (Sheet 1: Identified significant non-coding mutations sorted based on their scores, Sheet 2: Significant non-coding mutations based on empirical P-value). Some non-coding mutations that are bound by HepG2 cells TF with both actual and precise (100 base-pairs) sizes are shown in Table 1. The highest consistency was found to be 410. The second highest consistency at other genomic positions was 15, which is very less as compared to 410. It was also observed that the number of TFs binding at a specific location greatly change when the size of TFBS files was reduced to 100 base pairs. Table 1 also gives information about non-coding mutations that were found to be present within regions of Vista Enhancer Browser elements. It indicates that the mutations with smaller consistency were located at regions that show enhancer activity. It was analyzed that the mutation with consistency 4 was present within enhancer regions of the RCAN1 bracketing gene. This location is a TF binding site as well where 5 TF bind. Another mutation that was within the enhancer region of NDRG4 was bound by 7 TF.
Table 1.
Non-coding Mutations Identified at TF Binding Sites of HepG2 Cells a
| Genomic Location | Consistency | No. of TF Binding with Actual Size | No. of TF Binding with Precise Size | Names of TF Binding with Precise Size | Bracketing Gene in Vista Enhancer Browser |
|---|---|---|---|---|---|
| 5:1295113-1295113 | 410 | 4 | 1 | GABP | - |
| 22:40856967-40856967 | 15 | 13 | 3 | CJUN, ELF1, MAX | - |
| 5:1295046-1295046 | 11 | 6 | 5 | GABP, MAX, MXI1, POL2, SIN3AK20 | - |
| 4:24232389-24232389 | 10 | 19 | 4 | CEBPD, HDAC2, MAZ , SRF | - |
| 21:34544112-34544112 | 4 | 6 | 5 | MXI1, NFIC, P300, RAD21, SMC3 | RCAN1 |
| 16:58495226-58495226 | 2 | 11 | 7 | COREST, CTCF, HDAC2, MAFF, MAFK, RAD21, RFX5 | NDRG4 |
| 15:70099538-70099538 | 2 | 11 | 6 | ELF1, POL2, SIN3AK20, TAF1, TBP, YY1 | MIR629-UACA |
| 10:120851335-120851335 | 1 | 30 | 17 | BHLHE40, BRCA1, ELF1, FOSL2, FOXA1, FOXA2, GABP, HDAC2, MXI1, NFIC, RAD21, RFX5, RXRA, SIN3AK20,TAF1, TRF4, YY1 | - |
Abbreviation: TF, transcription factor.
a The column ‘Bracketing Gene in Enhancer Vista Browser’ provides names of genes showing enhancer activity where identified non-coding mutations were presenta.
4.2. Significance of Non-coding Mutations
The non-coding mutations were ranked based on their scores and P-values (Appendix in Supplementary File). Table 2 shows scores and P-values of some non-coding mutations. The highest score was found to be 5.385 out of 10 while the lowest score was 0.461. Some mutations in Table 2 were not highly consistent but still, they had high scores as they were bound by great numbers of TF while some mutations were consistent but only a few TF were binding there. Few mutations had similar scores but their consistency and number of TF’s binding were different. Many mutations in Table 2 are statistically significant as well (having a P-value less than 0.05). However, the P-value of a few mutations was above 0.05. It means those mutations are not significant.
Table 2.
Significance of Non-coding Mutations on the Basis of Their Scores and P-values a
| Genomic Locations | Consistency | No. of TF Binding with Precise Size | Scores (Out of 10) | P-Value (< 0.05) |
|---|---|---|---|---|
| 22:40856967-40856967 | 15 | 3 | 5.385 | 0.00175 |
| 20:17859269-17859269 | 1 | 39 | 5.333 | 0.5 |
| 6:157323527-157323527 | 3 | 32 | 5.102 | 0.00275 |
| 12:20815732-20815732 | 14 | 1 | 4.795 | 0.00695 |
| 20:49768490-49768490 | 1 | 34 | 4.692 | 0.5 |
| 18:58452573-58452573 | 13 | 1 | 4.461 | 0.00695 |
| 17:4278699-4278699 | 8 | 13 | 4.333 | 0.0001 |
| 2: 33013316-233013316 | 3 | 26 | 4.333 | 0.00275 |
| 14:24232389-24232389 | 10 | 4 | 3.846 | 0.001 |
| 14:39145540-39145540 | 7 | 8 | 3.358 | 0.0003 |
| 14:52873949-52873949 | 9 | 1 | 3.128 | 0.00695 |
| 17:75393912-75393912 | 7 | 4 | 2.846 | 0.00185 |
| 14:24425986-24425986 | 8 | 1 | 2.795 | 0.00695 |
| 5:72320307-72320307 | 2 | 10 | 1.948 | 0.0342 |
| 5:82351990-82351990 | 2 | 4 | 1.179 | 0.03505 |
| 8:84648494-84648494 | 1 | 1 | 0.461 | 0.5069 |
Abbreviation: TF, transcription factor.
a The scoring formula and calculations of P-values were based on the consistency of a particular mutation and the number of transcription factors (TF) binding there with precise size (100 base-pairs).
4.3. Association of Genes with Non-coding Mutations
Table 3 gives information about genes that were closest to non-coding mutations in greater numbers. It was found that 75 non-coding mutations were near the ALB gene. Some of them were in the upstream region while some were within the coding region of the ALB gene. The mutations were also reported in upstream and coding regions of the SYN3 gene. However, the mutations near MLLTP10P1,CNTNAP2,NPAS3, and LSAMP genes were in upstream, downstream, and coding regions. PLCB1, LINC00511, LINC01410, and WWOX genes had non-coding mutations in their downstream and coding regions. Some mutations occurred within coding regions of genes i.e., EYS, ZFHX3, PTPRN2, and AC0976344.
4.4. Mapping Non-coding Mutations to CTCF Binding Sites
The results of mapping with HepG2 cells' CTCF binding sites are shown in Table 4. A total of 49492 clusters were formed. It was observed that some clusters have a great number of non-coding mutations.
Table 4 shows that cluster number 48111 has the highest number of non-coding mutations i.e., 17. After that, 15 and 11 mutations are present in cluster numbers 17609 and 7433, respectively. Other important clusters (2565, 29170, and 32451) have 7, 6, and 6 mutations. In the majority of the clusters, the CTCF binding site did not lie between mutation and TSS of the gene. In two clusters, CTCF was found to be binding between all reported non-coding mutations and TSS of the gene.
4.5. Graphical Analysis of Significant Non-coding Mutations and Clusters
The selected genomic regions are graphically expressed in Figures 2 and 3. Figure 2 represents individual non-coding mutations, whereas Figure 3 represents clusters having a great number of non-coding mutations.
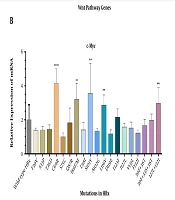
 Represents mutation 20:17859269-17859269, (B) Represents mutation 12:20815732-20815732. The red bars and blue bars in clinical variants represent copy number and gain. The green bar in ‘ClinVar Short Variant represents a benign clinical variant.")
Figure 2.
Graphical profiles of significant mutations (A) Represents mutation 20:17859269-17859269, (B) Represents mutation 12:20815732-20815732. The red bars and blue bars in clinical variants represent copy number and gain. The green bar in ‘ClinVar Short Variant represents a benign clinical variant.
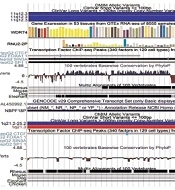
 Represents cluster 11:62841559-62841872, (B) Represents cluster 1:152018685-152018775. The Gencode v29 track displays basic genes present close to the given cluster. The Conservation tracks ‘Cons 100 Verts’ track and ‘Multiz Alignment of 100 vertebrates’ display regions that are conserved in multiple species in condensed form.")
Figure 3.
Graphical profiles of significant clusters (A) Represents cluster 11:62841559-62841872, (B) Represents cluster 1:152018685-152018775. The Gencode v29 track displays basic genes present close to the given cluster. The Conservation tracks ‘Cons 100 Verts’ track and ‘Multiz Alignment of 100 vertebrates’ display regions that are conserved in multiple species in condensed form.
All parts of Figures 2 and 3 indicate the presence of clinical variants at the given genomic regions. The red and blue bars indicate copy number variation. The bars are red for variants that experience loss of genetic material. The blue bars on the other hand represent the gain of genetic material. It means these regions are clinically significant as well. The genes expressed near these locations are also displayed. In Figure 3, some regions are also found to be conserved among different species. These conserved regions are generated from pair-wise alignments. The cluster shown in Figure 3B has CTCF binding, which is similar to the result shown in Table 4.
4.6. Analysis of Ras/MAPK Signaling Pathway
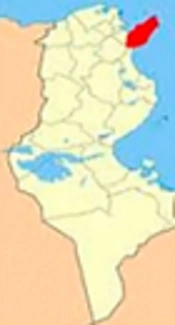
The non-coding mutations near genes that take part in the Ras/MAPK signaling pathway are shown in Figure 4. Figure is taken from the KEGG pathway database. Figure 4 shows mutations that are reported near most of the genes. The highest numbers of mutations were reported close to the PKC gene. Other genes with greater non-coding mutations near them include STAT3 and Grb2. There are a few genes, where the closest mutations were not reported i.e., Raf, MEK, CBP, and ELK1.
5. Discussion
The diseases like cancer can be prevented. The risk factors and causes of most cancers are known. Therefore, this knowledge can be used to avoid the majority of cancer-related deaths. In the case of liver cancer, viral hepatitis is the most common risk factor. It means the risk of developing liver cancer can be reduced when there is the active treatment of viral hepatitis. Today, only a small amount of these patients are successfully treated because of late diagnosis of disease. So, it is necessary to identify biomarkers for predicting liver cancer at its early stages. The main focus of this study is on non-coding mutations that occur in transcription factor binding sites of HepG2 cells.
Transcription factors are proteins that bind to the cis-regulatory elements. They regulate various cellular processes and control gene expression levels. If the mutations occur at binding sites of transcription factors, then, the binding of TFs to their sites will be disrupted. As a result, gene expression will be affected. The abnormal expression of the gene will, then, either enhance or reduce expression levels. Therefore, the mutations at TF binding sites can be termed driver mutations.
From Table 1, it is observed that 4 TFs were binding at a highly consistent location (5:1295113-1295113), but when the size was reduced, then, only 1 TF (GABP) was bound there. Similarly, 13 TFs were found to bind at another consistent location (22:40856967-40856967), but this number was reduced to 3 with the size reduction. It was also analyzed that enhancer regions where non-coding mutations were observed at TF binding sites were not highly consistent. Their consistency was 2, which means nucleotides bind randomly at TF binding sites. Therefore, these mutations may be considered random mutations. In Tables 1 and 2, a few mutations were not consistent but still, they were bound by the greater number of TFs with both actual and precise sizes and they had a high score as well. These mutations are very significant because if they occur in great numbers, they would surely cause disease. The significance of mutations can be inferred from the P-value. Some mutations were not statistically significant because we consider those alleles that were mutated at least once in case of consistency, whereas in the case of TF binding, the alleles with no TF binding were also considered along with TF bound alleles.
In a study by Li et al. (25), the authors discovered 11 novel driver genes through genome analysis of liver cancer. These genes include VAV3, TNRC6B, and RNF213. In Another study by Cleary et al. (26), the authors identified 13 new driver genes including TP53, CTNNB1, IGSF3, and ATAD3B. Hirotsu et al. in their study also identified mutations in TP53 and CTNNB1 (27). It shows that some of the genes indicated in Tables 3 and 4 are not identified as driver genes in liver cancer, but still, great numbers of non-coding mutations are reported near them. It implies that they may have some importance in liver cancer development. In Table 3, the closest distance from TSS of some genes was very less like in the case of the MLLTP10P1 gene; the mutation was reported at a distance of 60 base pairs from TSS. Similarly, the closest distance of mutations from TSS of WWOX, SYN3, and ALB genes was below 500 base pairs.
Table 3.
Genes Located Closest to the Non-coding Mutations in Great Numbers a
| Genes | Number of Non-coding Mutations Closer to Genes | Non-coding Mutations Present in the Upstream Region of Genes | Non-coding Mutations Present Within the Coding Region of Genes | Non-coding Mutations Present in Downstream Regions of Genes | The Closest Distance from Transcription Start Site (TSS) |
|---|---|---|---|---|---|
| ALB | 75 | 12 | 63 | 0 | 428 (up) |
| EYS | 43 | 0 | 43 | 0 | 0 |
| MLLT10P1 | 42 | 26 | 3 | 13 | 60 (down) |
| ZFHX3 | 38 | 0 | 38 | 0 | 0 |
| CNTNAP2 | 37 | 1 | 35 | 1 | 1220 (up) |
| LINC00511 | 36 | 0 | 32 | 4 | 5075 (down) |
| NPAS3 | 36 | 2 | 33 | 1 | 10540 (down) |
| WWOX | 35 | 0 | 33 | 2 | 276 (down) |
| PTPRN2 | 34 | 0 | 34 | 0 | 0 |
| LINC01410 | 32 | 0 | 24 | 8 | 673 (down) |
| LSAMP | 32 | 4 | 26 | 2 | 4801 (up) |
| PLCB1 | 32 | 0 | 31 | 1 | 3472 (down) |
| SYN3 | 27 | 4 | 23 | 0 | 363 (up) |
a The terms ‘up’ and ‘down’ represent upstream and downstream regions of genes. The closest distance from transcription start site (TSS) was written as 0 for mutations present within coding regions of genes.
Table 4.
Mapping of Clusters Having a Great Number of Non-coding Mutations with CTCF Binding Sites of HepG2 Cells a
| Cluster No. | Cluster Size | No. of Mutations in a Cluster | Highest Score in Cluster | Closer Genes | Closest Distance from TSS | CTCF Binding Between Gene TSS and Mutation |
|---|---|---|---|---|---|---|
| 48111 | 9:62802442-62802699 | 17 | 1.987 | LINC01410 | 0 (within) | 12 No |
| 17609 | 17:8173337-8173599 | 15 | 3.282 | TMEM107, SNORD118 | 0 (within), 11 (upstream) | 15 No, 15 No |
| 7433 | 11:62841559-62841872 | 11 | 5.333 | WDR74, RNU2-2P | 50 (upstream), 27 (downstream) | 9 No, 9 No |
| 2565 | 1:152018685-152018775 | 7 | 1.589 | AL450992.1, NBPF18P | 0 (within), 0 (within) | 7 Yes, 7 Yes |
| 29170 | 20: 53941417-53941434 | 6 | 2.589 | BCAS1, AC005220.1 | 0 (within), 0 (within) | 6 No, 6 No |
| 32451 | 3:113051365-113051399 | 6 | 1.307 | AC078785.1, AC078785.2 | 0 (within), 0 (within) | 6 Yes, 6 Yes |
Abbreviation: TSS, transcription start site.
a ‘Yes’ is written when CTCF binds between mutation and TSS gene and vice versa.
It has been found in approximately 70% of cases that the regulatory region of a gene lies within 100 kb (28). The coding region of one gene can be a regulatory region for another gene. Therefore, those mutations that are reported within coding regions of some genes are significant as well. They may be coding for TSS genes and non-coding for any gene present in the upstream/downstream region. In Table 4, the clusters, where CTCF binding sites were not present between mutation and TSS, might be considered regulatory regions of corresponding genes. So, the mutations reported in these regulatory regions are highly significant as they may have potential to the drive disease. However, the clusters, where CTCF was binding between TSS and mutations, cannot be regarded as regulatory regions for the particular genes.
Figure 2 shows that the mutation at location 20:17859269-17859269 has no gene expression, whereas the SLCO1B3 gene is expressed at location 12:20815732-20815732. In Figure 3, the regions that are found to be conserved among different species can be mutated in those species as well. There are more conserved regions in Figure 3A compared to 3B. The bars with the TF of HepG2 cells are displayed only when the corresponding TF binds there. The darkness of bars for TF of HepG2 cells represent locations that are enriched with specific TF. CTCF binding in cluster 1:152018685-152018775 shows that the mutations reported in that region are not in regulatory regions of specific genes. Figures 2 and 3 validated the acquired results. It indicates that the regions selected for graphical analysis have great importance and can be considered epigenetic markers for predicting liver cancer. However, detailed analysis is required for better understanding.
Ras, Raf, MEK, and ERK are signaling molecules in the Ras/MAPK signaling pathway. These molecules activate this pathway, which results in gene transcription; the transcribed genes code for proteins that are involved in cellular growth and proliferation. Figure 4 indicates that no non-coding mutation is reported near Raf and MEK molecules, but they might have coding mutations.

Figure 4.
Analysis of Ras/MAFK signaling pathway taken from KEGG pathway. The numbers of mutations that occurred near genes are mentioned in red beside gene names.
5.1. Conclusion
The present study provides a comprehensive analysis of non-coding mutations through bioinformatics tools. The identification of recurrent/consistent somatic mutations at TF binding sites in non-coding variants suggests that they may play a significant role in driving Hepatocellular Carcinoma (HCC). This information will help analyze non-coding regions contributing to the development of liver cancer. The results of this study are also essential in designing appropriate research strategies. This is because mutations in non-coding regions are more likely to affect the regulatory elements of genes. They may also cause structural variations in genes resulting in gene disruptions. The identified pathogenic alleles can be considered novel biomarkers for liver cancer diagnosis and prognosis. They may also act as therapeutic targets for the treatment of liver cancer. However, further assessment is required for confirmation of the acquired results.