


1. Background
Myopia is one of the most common eye disorders globally (1). Researchers have estimated that by the year 2050, approximately ~49.8% and 9.8% of individuals all over the world will develop myopia and high myopia (HM) (2). Furthermore, myopia increases the risk of developing a lot of eye diseases, including cataracts, retinal dysfunction, and myopic maculopathy (3, 4). Nevertheless, the underlying molecular mechanisms are still largely undetermined.
Myopia is a complex disease that is associated with both genetic and environmental factors, such as long-time reading and close work, which are believed to be associated with oxidative stress (OS) (5-7). In fact, OS is a condition associated with an imbalance between overproduction of reactive oxygen species (ROS) and reduced antioxidant capacity (8). Because mitochondria are the main sources for ROS generation and clearance, mutations/variants in mitochondrial DNA (mtDNA) will cause a defect in ATP synthesis and an increase in oxygen radicals (9). Furthermore, Wang et al. found a potential association between mtDNA genetic background and the phenotypic manifestation of myopia in the Chinese population (10), emphasizing the important role of mtDNA mutations/variants in myopia. Nevertheless, currently, there are no studies regarding the association between mitochondrial tRNA (mt-tRNA) mutations and HM.
2. Objectives
The objective of our study was to analyze the spectrum of mt-tRNA mutations/variants in 150 children with HM and 100 healthy controls from Dongguan City Maternal & Child Health Hospital by using PCR and direct sequencing. Moreover, to assess the pathogenicity of mt-tRNA variants, the phylogenetic conservation and pathogenicity scoring system were employed to evaluate these mt-tRNA mutations/variants.
3. Methods
3.1. Subjects
From January 2018 to January 2020, a total of 150 myopic children (60 boys and 90 girls) younger than 16 years, together with 100 healthy children (50 boys and 50 girls) younger than 15 years who had normal vision, were recruited from the Department of Ophthalmology, Dongguan City Maternal & Child Health Hospital. The age of HM patients ranged from seven to 16 years, with an average of 11 years, and the age of healthy children ranged from eight to 14 years, with an average of 10 years.
The Visual Acuity (VA) was carried out to examine HM. Then, HM was classified as spherical equivalent ≤ -5.00 D and axial length ≥ 26.5 mm, according to a previous study (11). We excluded the patients if they had other eye or relevant systemic diseases. In addition, 100 children with normal vision were enrolled based on the following criteria: (1) Born in Dongguan City of Guangdong Province, (2) best-corrected VA ≥ 1.0, (3) no other known eye or relevant systemic diseases that could interfere with the results, and (4) no family history of myopia.
All procedures were performed as per the Declaration of Helsinki. The Ethics Committee of Dongguan City Maternal & Child Health Hospital approved the study. Written informed consent was obtained from all participants.
3.2. Screening for mt-tRNA Variants
In order to see the spectrum of mt-tRNA variants, PCR-Sanger sequencing was performed. First, the genomic DNA of 150 subjects with HM and 100 controls was isolated from blood samples using the Puregene DNA extraction Kit (Qiagen, Valencia, CA). Then, the DNA concentrations and purity were measured by ultraviolet-visible spectrophotometry.
The PCR products spanning the entire mt-tRNA genes were amplified in all participants using primer sequences described in a previous study (12). Subsequently, the PCR products were purified and sequenced by an ABI automated DNA sequencer. Sequences were edited using DNAStar software (DNASTAR Inc., Madison, WI, USA), and the mtDNA variants were scored relative to the revised Cambridge Reference sequence (rCRS, GenBank Accessible No: NC_012920.1) (13).
3.3. Structural Analysis
Stem and loop structures were defined based on published human mt-tRNA secondary structures (mammalian mt-tRNA database: http://mamit-trna.u-strasbg.fr/) (14), with tertiary structure interactions for these tRNA molecules being determined by referring to the relevant literature (15).
3.4. Conservation Assessment
An interspecies analysis was conducted by comparing mtDNA sequences across 16 different vertebrate species (http://trna.bioinf.uni-leipzig.de/DataOutput/), as described previously (16). These species were as follows: Cebus albifrons, Colobus guereza, Gorilla gorilla, Homo sapiens, Hylobates lar, Lemur catta, Macaca mulatta, Macaca sylvanus, Nycticebus coucang, Pan paniscus, Pan troglodytes, Pongo pygmaeus, Pongo abelii, Papio hamadryas, Tarsius bancanus, and Trachypithecus obscurus. We further calculated the conservation index (CI) by comparing the human mtDNA variant with the other 15 species involved in this study (17).
3.5. Determining Pathogenicity
Previously, Yarham et al. generated a weighting scoring system that could be used to determine the pathogenicity of an mt-tRNA variant (18). According to their standard, if the total score was less than 6 points, it belonged to “neutral polymorphism”; if the score was 7-10 points, it was classified as “possible pathogenic”, and if the score was more than 11 points, it was regarded as “definitely pathogenic”.
4. Results
4.1. Mutational Screening for HM-related mt-tRNA Variants
We carried out a genetic screening program for HM-associated mt-tRNA variants. The PCR and direct sequencing analysis revealed six possibly pathogenic mutations: tRNALeu (UUR) T3290C, tRNAIle A4317G, tRNAAla G5591A, tRNASer (UCN) T7501C, tRNAHis T12201C, and tRNAThr G15915A. Among these sequence variants, the T3290C, A4317G, and G5591A variants were homoplasmic, whereas the T7501C, T12201C, and G15915A variants were heteroplasmic. Furthermore, the T3290C variant occurred in two out of 150 myopic children (1.33%), the A4317G variant in one patient with HM (0.67%), the G5591A variant in one child with HM (0.67%), the T7501C variant in two children with HM (1.33%), the T12201C variant in one out of 150 myopic children (0.67%), and the G15915A variant in one out of 150 myopic children (0.67%). However, we did not find any mt-tRNA variants in control subjects. The characteristics of six HM-associated mt-tRNA variants are listed in Table 1.
| tRNA Species | Sequence Alteration | Homoplasmy/ Heteroplasmy | Location | Numbering In trna | CI (%) | No. of 150 Myopic Children (%) | No. of 100 Controls (%) | Disease Association |
|---|---|---|---|---|---|---|---|---|
| tRNALeu(UUR) | T3290C | Homoplasmy | TψC loop | 59 | 100 | 2 (1.33) | 0 | Diabetes; hypertension |
| tRNAIle | A4317G | Homoplasmy | TψC loop | 59 | 100 | 1 (0.67) | 0 | Deafness; cardiomyopathy |
| tRNAAla | G5591A | Homoplasmy | Acceptor arm | 69 | 100 | 1 (0.67) | 0 | Myopathy |
| tRNASer(UCN) | T7501C | Heteroplasmy | DHU loop | 15 | 100 | 2 (1.33) | 0 | Cardiovascular disease |
| tRNAHis | T12201C | Heteroplasmy | Acceptor arm | 68 | 100 | 1 (0.67) | 0 | Deafness |
| tRNAThr | G15915A | Heteroplasmy | Anticodon stem | 28 | 100 | 1 (0.67) | 0 | Encephalomyopathy |
Molecular Features of HM-associated mt-tRNA Variants
4.2. Evaluation of mt-tRNA Variants
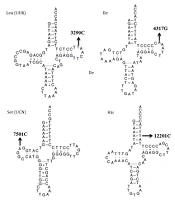
We next assessed the potential pathogenicity of these mt-tRNA variants by using the following criteria: (1) CI value > 75%, consistent with evolutionary conservation at a given locus, as suggested by Ruiz-Pesini and Wallace (17), (2) being present in < 1% of control patients, and (3) capable of making alterations in the structure and/or function of mt-tRNA molecules. As shown in Table 1, all of the six mt-tRNA variants identified were well conserved between various species (CI = 100% for all). In addition, none of them were found in control subjects. The locations of these variants within tRNA secondary structures are shown in Figure 1. Typically, mt-tRNA molecules had a clover-like morphology with acceptor arm, anticodon stem, and TψC loop and DHU loop (19, 20). Of these variants, two occurred at the acceptor arm, two were localized at the TψC loop, one occurred at the DHU loop, and one occurred at the anticodon stem. Based on the analysis, these six evolutionarily conserved tRNA variants were predicted to disrupt the tRNA structure and function.

The mt-tRNA variants
4.3. Determining Pathogenicity of Candidate mt-tRNA Variants
To evaluate the pathogenic role of an mt-tRNA variant, the pathogenicity scoring system was used (18). Therefore, we found that the total scores of T3290C, A4317G, G5591A, T7501C, T12201C, and G15915A variants were 7, 15, 15, 7, 17, and 11 points, respectively, belonging to “possibly pathogenic” and “definitely pathogenic” (Table 2).
| Scoring Criteria | T3290C Variant | Score | A4317G Variant | Score | G5591A Variant | Score | T7501C Variant | Score | T12201C Variant | Score | G15915A Variant | Score | Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| More than one independent report | Yes | 2 | Yes | 2 | Yes | 2 | Yes | 2 | Yes | 2 | Yes | 2 | ≤ 6 points: neutral polymorphisms 7~10 points: possibly pathogenic; 11 - 13 points (not including evidence from single fiber, steady-state level, or trans-mitochondrial cybrid studies): probably pathogenic; ≥ 11 points (including evidence from single fiber, steady-state level or trans-mitochondrial cybrid studies): definitely pathogenic |
| Evolutionary conservation of the base pair | Multiple changes | 0 | No change | 2 | No change | 2 | No change | 2 | No change | 2 | No change | 2 | |
| Variant heteroplasmy | No | 0 | No | 0 | No | 0 | No | 0 | Yes | 2 | No | 0 | |
| Segregation of the mutation with disease | Yes | 2 | Yes | 2 | Yes | 2 | No | 0 | Yes | 2 | Yes | 2 | |
| Histochemical evidence of mitochondrial disease | No evidence | 0 | Strong evidence | 2 | Strong evidence | 2 | No evidence | 0 | Strong evidence | 2 | No evidence | 0 | |
| Biochemical defect in complex I, III or IV | No | 0 | Yes | 2 | Yes | 2 | No | 0 | Yes | 2 | No | 0 | |
| Evidence of mutation segregation with biochemical defect from single-fiber studies | No | 0 | No | 0 | No | 0 | No | 0 | No | 0 | No | 0 | |
| Mutant mt-tRNA steady-state level or evidence of pathogenicity in trans-mitochondrial cybrid studies | Weak evidence | 3 | Strong evidence | 5 | Strong evidence | 5 | Weak evidence | 3 | Strong evidence | 5 | Strong evidence | 5 | |
| Maximum score | Possibly pathogenic | 7 | Definitely pathogenic | 15 | Definitely pathogenic | 15 | Possibly pathogenic | 7 | Definitely pathogenic | 17 | Definitely pathogenic | 11 |
Predicted Pathogenicity of HM-associated mt-tRNA Variants
5. Discussion
Since human mtDNA codes 13 polypeptides that are essential for oxidative phosphorylation (OXPHOS), it generates ROS as a toxic byproduct (21). The overproduction of ROS may have serious consequences such as damaging lipids, proteins, and DNA or RNA, increasing OS (22, 23). In fact, the retina is very sensitive to be influenced by ROS because it needs high levels of oxygen consumption (24). Therefore, we hypothesized that mtDNA mutations or variants may lead to mitochondrial dysfunction, and play a putative role in the pathogenesis of HM.
For this purpose, the frequencies of mt-tRNA variants in 150 children with HM and 100 control subjects were analyzed by direct sequencing. As a result, we identified six possible pathogenic mt-tRNA variants: tRNALeu (UUR) T3290C, tRNAIle A4317G, tRNAAla G5591A, tRNASer (UCN) T7501C, tRNAHis T12201C, and tRNAThr G15915A, which were not detected in 100 controls. Among them, the homoplasmic T3290C variant that occurred at position 59 in the TψC loop of tRNALeu (UUR), was regarded as a risk factor for hypertension (25). Moreover, the A4317G variant affected a very conserved adenine at position 59 in the T-loop of tRNAIle. This variant, however, introduced a novel Watson-Crick base-pairing (59G-54C) and led to the re-arrangement of the TψC loop region (26, 27). A recent experimental study revealed that the A4317G variant influenced the steady-state and aminoacylation efficiency of tRNAIle, and aggravated the defective mitochondrial translation and respiratory phenotypes associated with the 12S rRNA A1555G mutation (28). In addition, the G to A transition at position 5591 was found to be associated with myopathy (29). Structurally, the G5591A variant disrupted the very conserved Watson-Crick base-pairing (4G-69C). Furthermore, the G5591A variant was localized at the acceptor arm in the 3’-end of tRNAAla, which was crucial for tRNA structure and function (30). Thus, it can be speculated that the G5591A variant may influence the tRNA metabolism and lead to mitochondrial dysfunction.
Moreover, the T7501C variant was localized at the DHU loop of tRNASer (UCN) (position 15) with heteroplasmy form. The nucleotide at position 15 was extremely conserved from various species. Bioinformatics analysis indicated that the T7501C variant can alter the secondary structure of tRNASer (UCN) and may result in a failure in mt-tRNA metabolism (31-33). While the T12201C variant, which is located at the acceptor arm of tRNAHis, abolished a well-conserved base-pairing (5A-68T), functional analysis of cybrid cells containing this variant revealed a significant reduction of tRNAHis stability level (34-36). Therefore, this variant may impair the tRNA metabolism, which is responsible for mitochondrial dysfunction. In particular, the T12201C variant reduced the OXPHOS-related polypeptides, as evidenced by a recent study (35). Furthermore, the G15915A variant disrupted a classic Watson-Crick base-pairing in tRNAThr, which was regarded as a pathogenic mutation associated with mitochondrial encephalomyopathies (37, 38). The alteration in tRNA structure may impair tRNAThr functions, subsequently affecting the mitochondrial protein translation, which was similar to the tRNALys A8344G variant (39).
Based on these observations, we proposed that the possible molecular mechanisms underlying the mt-tRNA variants for HM may be as follows. First, the variant itself alters the secondary structure of the corresponding tRNA and causes a failure in tRNA metabolisms, such as affecting the steady-state level, aminoacylation ability, and post-transcriptional modification. Defects in tRNA metabolism will lead to the impairment of mitochondrial protein translation and respiratory chain function. As a result, these events will cause mitochondrial dysfunction, including increased ROS production and decreased ATP synthesis. Subsequently, OS occurs due to an imbalance between ROS and antioxidants, potentially involved in the pathogenesis of HM.
In conclusion, our study indicated that mt-tRNA variants may play important roles in the pathogenesis of HM. Screening for common mt-tRNA variants is advised for the diagnosis of children with HM.
