




1. Introduction
GATA 2 deficiency syndrome is an autosomal dominant genetic disease caused by a defect in GATA 2, which is an essential transcription factor in the development of embryogenesis, hematopoiesis, and lymphatic angiogenesis. Initially, this syndrome was known as MonoMAC and dendritic cell, monocyte, and B and NK lymphoid (DCML) deficiency. The symptoms of this syndrome include disseminated non-tuberculosis mycobacterial (NTM) infections, susceptibility to viral infections, profound monocytopenia, and B-cell and NK cell lymphopenias. The presentation age of the disease is typically late childhood, but the earliest reported age of onset of the symptoms is 14 months (1, 2). Furthermore, since GATA-2 is a hematopoietic transcription factor, patients with this syndrome are prone to acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) (3). The prevalence of MDS in these patients is 21% (1), particularly childhood myelodysplastic syndrome (cMDS) with hypoplastic bone marrow and somatic monosomy 7 (4). This case report describes a patient who presented with pyoderma gangrenosum (PG). Ultimately, the diagnosis of cMDS was made, occurring in the context of an underlying GATA2 deficiency syndrome.
2. Case Presentation
The patient is an 11-year-old female with normal growth parameters. Her parents are healthy and unrelated; she had normal development, and there is no history of recurrent infections or lymphedema. However, according to her parents, during the COVID-19 outbreak, she was hospitalized due to severe diarrhea. Mild persistent thrombocytopenia and a mildly decreased hemoglobin (Hb) level were also noted before her current presentation. She presented with a small red nodule on her face in April 2022 (Figure 1A). In the first two months, due to the small size of the lesion, the parents did not seek medical attention, and no treatment was administered. After that, the rash grew rapidly, and the patient underwent local and traditional treatments (Figure 1B). However, these were not effective, so a dermatologist evaluated the patient, and a lesion biopsy along with various tests were performed to rule out infectious causes such as tuberculosis, Leishmania, and fungus (Figure 1C). All the results were negative. An ultrasound examination of the facial soft tissue revealed a hypoechoic lesion measuring 15 × 6 mm with increased vascularity, a maximum arterial velocity of 6 cm/s, and RI = 0.5, with a vascular lesion or malignancy mentioned in the differential diagnosis.

A, face skin lesion at presentation; B, the lesion after growing; C, the lesion after biopsy.
A complete blood cell count (CBC) test at this time revealed moderate thrombocytopenia with a platelet count of 75×109/L. Red blood cell (RBC) and white blood cell (WBC) counts were normal, and Hb was at the lower limit of normal, measuring 11.6 g/dL. Due to the lack of diagnosis, an immunologist evaluated the patient to rule out immunodeficiency disorders. The IgG immunoglobulin level was mildly increased, and the number of B and T cells was normal. A marked decrease in the CD4 to CD8 ratio was observed, with a value of 0.2. The Nitro blue tetrazolium (NBT) test result was also normal. In parallel with the tests (two months later), a similar lesion appeared on the leg skin and started to grow. The ulcer was a large ulcerative and necrotic plaque with a violaceous border. A second biopsy was performed from this area (Figure 2A and B). The skin biopsy of the leg lesion revealed a dense mixed inflammatory reaction in the dermis with vague suppurative granuloma formation and fistulation. Special histochemical staining and polymerase chain reaction (PCR) tests for Mycobacterium tuberculosis and Leishmania were negative.
 was made.")
A, leg skin lesion at first; B, the lesion at the time that diagnosis of myelodysplastic syndrome (MDS) was made.
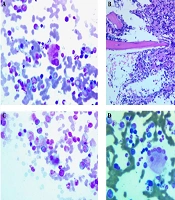
The diagnosis was not confirmed yet. However, two months of empirical antifungal treatment with fluconazole was started for the patient, which was not effective. Meanwhile, new hematological and immunological tests were requested for the patient (four months after the onset of symptoms). In the CBC test, a prominent reduction in platelets was observed at the level of 50 × 109/L, and the number of WBCs was at the upper limit of normal. At this time, PG was finally proposed for the patient, and the investigation was started to find the underlying causes. Due to the positive family history of inflammatory bowel disease (IBD), a colonoscopy was performed for the patient, which was normal, and a bone marrow biopsy was performed. The biopsy revealed 80% cellularity with 7% blasts and prominent dysmegakaryopoiesis (megakaryocytes with multiple separated nuclei) (Figure 3A), (micromegakaryocyte in bone marrow biopsy) (Figure 3B), (small binucleated megakaryocyte) (Figure 3C), (megakaryocytes with many round separate nuclei) (Figure 3D), (about 40%), mild dyserythropoiesis, and dysgranulopoiesis (10%).
; B, micromegakaryocyte in bone marrow biopsy (H&E staining 400X); C, small binucleated megakaryocyte (Giemsa staining 1000X); D, megakaryocytes with many round separate nuclei (Giemsa staining 1000X).")
A, megakaryocyte with multiple separated nuclei (Giemsa staining 1000X); B, micromegakaryocyte in bone marrow biopsy (H&E staining 400X); C, small binucleated megakaryocyte (Giemsa staining 1000X); D, megakaryocytes with many round separate nuclei (Giemsa staining 1000X).
At this time, the platelet count was severely decreased to 20 × 109/L. A bone marrow cytogenetic study revealed monosomy 7, which completed the diagnostic criteria for MDS (Figure 4). According to the new WHO classification of hematologic malignancy, the patient was classified as childhood myelodysplastic syndrome with increased blasts (cMDS-IB). After that, since cMDS usually occurs in the context of inherited bone marrow failure disorders (5, 6), a genetic test (whole exome sequencing) was requested for the patient, and a heterozygous likely pathogenic variant was identified in the GATA2 gene (c.G880P, P: E294*) with autosomal dominant inheritance. The patient was a candidate for bone marrow transplantation, and the necessary clinical and paraclinical examinations were started to find a suitable donor. Unfortunately, MDS transformed into AML about four months after the diagnosis of MDS. Bone marrow transplantation was performed two months later. Fortunately, the patient's bone marrow transplant resulted in full donor chimerism. The patient has gastrointestinal graft-versus-host disease (GVHD) and is currently under treatment.

Chromosomal analysis revealed abnormal female chromosomal complement with monosomy 7 in all analyzable cells.
3. Discussion
The most noteworthy aspect of this case is the presentation of PG followed by MDS as initial manifestations of GATA2 deficiency syndrome — a rare and under-recognized condition. To our knowledge, this specific sequence of clinical events has not been previously reported in the literature. GATA2 is a critical transcription factor involved in hematopoiesis, regulating gene expression in hematopoietic stem and progenitor cells. It is predominantly expressed in precursor cells of the bone marrow, including megakaryocytes, erythroid progenitors, and mast cells (7, 8). GATA2 deficiency leads to a spectrum of clinical manifestations due to immune dysregulation, bone marrow failure, and impaired hematopoietic lineage development. One of the characteristic immunological features observed in affected patients is an inverted CD4/CD8 T-cell ratio, as seen in our patient (9).
The GATA2 gene, located on chromosome 3, demonstrates autosomal dominant inheritance with incomplete penetrance. Approximately 62% of mutations are germline, while 38% are de novo (10). Mutations in this gene predispose patients to both hematologic conditions — such as MDS and AML — and non-hematologic disorders, including immunodeficiency (MonoMAC syndrome), lymphedema, and thrombotic events (MIM# 601626, 614286, 614038, 614172; MONDO:0042982).
In a study by Wlodarski et al., 72% of adolescents diagnosed with MDS and monosomy 7 were found to have GATA2 mutations (11). Similarly, Nell et al. reported a pediatric patient with GATA2 deficiency that presented with pancytopenia and disseminated NTM infection secondary to MDS (10). In our case, the patient presented at the age of 11, consistent with the typical age of symptom onset in GATA2 deficiency syndrome. The phenotypic expression of the disease is highly variable, ranging from recurrent infections in early life to mild autoimmune manifestations, or in some cases, asymptomatic individuals well into adulthood (12). Our patient had no significant history of recurrent infections, aside from a single hospitalization for severe diarrhea, likely due to COVID-19 at age nine. Interestingly, the first clinical manifestation was a small ulcerative lesion on the face, which subsequently progressed to involve the leg and oral mucosa. Histopathological and microbiological investigations eventually led to a diagnosis of PG — an atypical initial presentation of GATA2 deficiency, not previously described in available medical literature. The likely reason for the absence of similar case reports is the inherent rarity of MDS in children, combined with the uncommon presentation of PG as an initial manifestation. Furthermore, genetic testing is not routinely performed in pediatric MDS, which may contribute to the underdiagnosis of underlying genetic syndromes such as GATA2 deficiency.
Genetic analysis identified a heterozygous GATA2 mutation in our patient (c.G880T, NM_032638.5), resulting in a premature stop codon (P: E294*). Although this specific mutation has not been reported in ClinVar, it is considered likely pathogenic based on predictive algorithms in the Franklin GENOX database. To date, over 179 pathogenic or likely pathogenic GATA2 variants have been reported, most of which are loss-of-function mutations, including frameshifts (40%), nonsense mutations (10.1%), splice-site alterations (6.7%), and gene deletions (15%). A smaller proportion of cases involve missense mutations, often affecting the zinc finger 2 (ZF2) domain (13).
The PG is a rare neutrophilic dermatosis with a pathogenesis that is not fully understood. Proposed mechanisms include neutrophil dysfunction, autoimmune mechanisms, genetic predisposition, and environmental triggers (14). The PG is typically a diagnosis of exclusion, and in approximately 50% of cases, it is associated with underlying systemic diseases such as IBD, autoimmune arthritis, or hematologic disorders (15).
Among these, MDS is a particularly important underlying condition. The MDS is a clonal stem cell disorder marked by ineffective hematopoiesis and has been increasingly recognized as a potential trigger for PG (16). The association may be due to immune dysregulation in MDS, especially involving aberrant neutrophil function and autoimmune activity. Neutrophil dysfunction in PG — encompassing defective chemotaxis, phagocytosis, and migration — can be exacerbated in the context of MDS. Furthermore, genetic and immunological abnormalities inherent in MDS may contribute to sustained inflammation and skin involvement. T-cell dysregulation and B-cell-mediated autoantibody production against skin antigens have also been implicated, and PG in the setting of MDS is considered a marker of poor prognosis and potential leukemic progression (17).
In our patient, the diagnostic process for PG was protracted, taking approximately six months from initial referral to diagnosis. Multiple histopathological, molecular, and microbiological evaluations were performed to exclude infectious causes such as fungal infections, tuberculosis, and leishmaniasis. This delay reflects the diagnostic challenge of PG, which lacks specific histological findings and relies heavily on the exclusion of other causes.
Furthermore, the diagnosis of MDS in children is inherently rare (6), which contributed to the initial diagnostic uncertainty. However, the presence of unexplained cytopenia, particularly thrombocytopenia and a reversed CD4/CD8 ratio, should have prompted earlier hematologic evaluation, including a bone marrow biopsy. This delay is clinically significant, as early diagnosis and intervention are essential in cMDS to prevent progression to AML, especially in patients with high-risk cytogenetic abnormalities. Moreover, PG in the context of MDS is associated with an unfavorable prognosis and may be indicative of disease acceleration (18).
3.1. Conclusions
This case highlights the rare presentation of PG as the initial manifestation of GATA2 deficiency syndrome complicated by cMDS. It underscores the importance of considering underlying genetic and hematologic disorders in pediatric patients presenting with refractory neutrophilic dermatoses and unexplained cytopenias. Early recognition and comprehensive evaluation, including genetic testing, are crucial for timely diagnosis and management, which may improve clinical outcomes and guide prognostication in this vulnerable population.
