
Fulltext
Hepatic mesenchymal hamartoma (HMH) is an uncommon benign tumor of childhood. Typically a patient will present with an enlarged, nontender abdominal mass. No specific panel of laboratory tests is characteristic of HMH. Laboratory studies often reveal normal liver function tests and various tumor markers, including human chorionic gonadotropin, alpha fetoprotein, and vanillylmandelic acid, are usually negative. Abdominal ultrasound can demonstrate either a multicystic or solid mass[1]. Although not required for diagnosis, computed tomography and magnetic resonance imaging are useful for surgical planning. Surgery, consisting of either enucleation or lobectomy, traditionally has been the treatment of choice in the postpartum period; however, less invasive techniques, such as laparoscopic fenestration have also been used successfully[2]. Because the natural course of this tumor is initially rapid increase and subsequently decrease in size, some investigators have opted for ‘‘watchful waiting’’ in asymptomatic patients[3].
At 29 weeks of gestation, a 960g male infant presented with distress and abdominal distention during the postnatal period. There was visible fullness in the right upper quadrant of the abdomen. The liver was palpable, 6 cm below the left subcostal margin in the mid-clavicular line. Serum bilirubin was 3.91 mg/dl, SGPT 56 U/L, SGOT 8U/L, blood urea 21 mg/dl, Hb 14g. Serum alpha-fetoprotein was in normal range. Bleeding, clotting, and prothrombin time, as well as platelet count and blood gas analysis were normal. X-ray of the chest showed elevated right dome of the diaphragm. US of the abdomen revealed a large, 8.8×7.0×4.7 cm sized anechoic cystic mass occupying almost the entire left lobe and superior segment of right lobe of the liver. There were multiple internal septations and multilobules within the mass. However, no solid component was visible. Other hepatic parenchymal echoes were homogeneous. The right lobe of the liver was enlarged. The portal vein showed normal flow. No portal or retroperitoneal lymphadenopathy was seen and there was minimal perihepatic ascites. On first MRI scan, the lesion was 4.7×5.3×5.6 cm, hyper intense on T2W image and hypo intense on T1W images confirming its cystic nature in the right lobe (Fig 1). Control ultrasound and MRI scan was repeated 3 months later. Anechoic cystic mass size was 10.8×6.4×5.6 cm. MRI scan did not show enlargement of the previously diagnosed cystic lesions but gave rise to multilocular
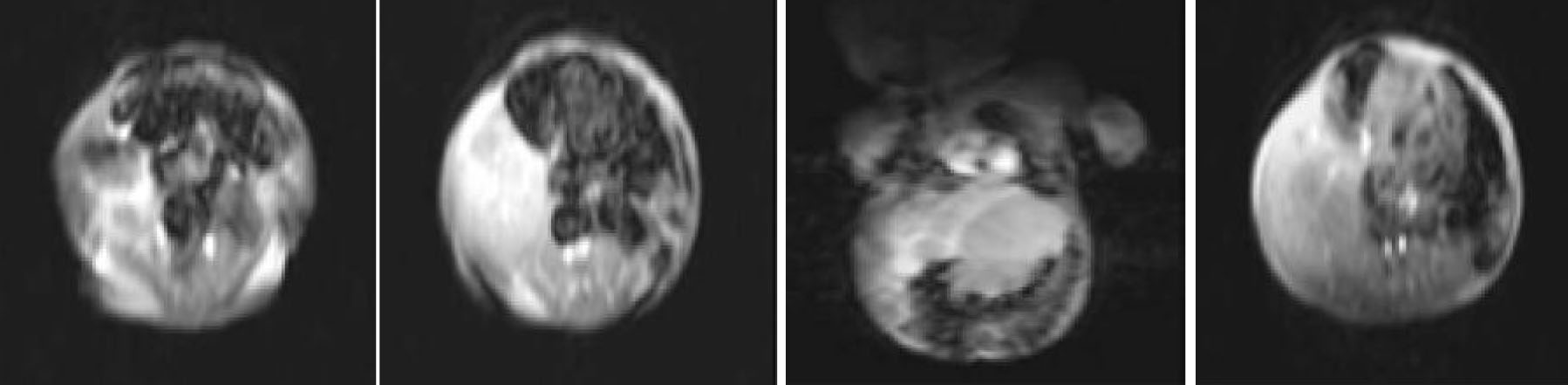
Fig. 1: MRI in the age of 1-month-old showing a large, dominantly cystic hepatic mesenchymal hamartoma
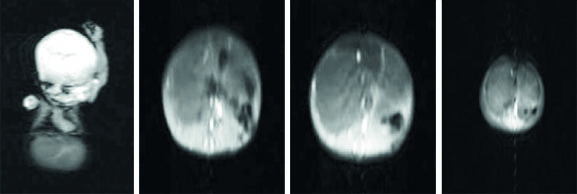
Fig. 2: Second MRI in the age of 3-months with dominantly cystic hepatic mesenchymal hamartoma
septations shown in the second MRI (Fig 2). The diagnosis was established by ultrasonograpy and MRI scan, and histologically confirmed. Supportive therapy, such as mechanical ventilation, fluid therapy and enteral nutrition was given to preterm infant admitted to neonatal intensive care. The case was not eligible for surgical operation of the liver mass. Follow up showed no complications and liver mass had not changed in size. Regarding the good condition, the patient was discharged from NICU.
The typical HMH presentation is one of asymptomatic, rapid abdominal distention with a palpable mass on physical examination[4]. The tumor can cause respiratory distress or apnea[5]. Our case presented with abdominal distention and respiratory distress in early postnatal period. Usually liver function tests are normal and alpha-fetoprotein is elevated, which is believed to be secreted by the proliferating hepatocytes within the tumor[4]. In our case liver function tests and coagulation tests were normal. The radiological appearance is one of a large, uni- or multi-cystic, avascular mass occupying part of the liver[4]. Ultrasound of the abdomen reveals a large, anechoic cystic mass of the liver. There are multiple internal septations and multilobules within the mass but no solid component is visible. MRI scan demonstrates the complex nature of the cystic mass. In our case ultrasound scan and MR scan showed multiple internal septations and multilobules within the mass at the first and 3rd month of life.
HMHs are best treated by complete excision. Excision may be by conventional hepatic resection or by non-anatomical excision with a small margin of normal liver. If the patient is in satisfactory clinical condition and if the lesion is resectable, the prognosis is excellent. Neonates generally have a better outcome than fetuses. Pedunculated lesions are amenable to laparoscopic resection[6]. In previously reported cases of mesenchymal hamartoma occupying part of the liver, the tumors were successfully treated by surgical resection, and the remaining liver tissue provided adequate liver function[5,6]. This treatment was considered inadequate for our case due to prematurity of the infant.
Nonoperative management may be appropriate in selected cases (e.g., infants with a biopsy-proven HMH and a prominent vascular component). As far as we know, there were a few cases of spontaneous regression in preterm neonates reported in literature. Barnhart et al[3] observed spontaneous regression of asymptomatic HMH, and suggest non-operative management for asymptomatic patients. In addition, nine patients in whom HMH showed a variable degree of spontaneous regression have been reported: four dominantly solid tumors were highly vascular, three of which became heavily calcified as the tumor regressed; this suggests that HMHs with a prominent angiomatous component are more likely to regress. None of the lesions disappeared completely, and in all but one the follow-up observation lasted less than 3 years[3,7]. In our case, the HMH was characterized by angiomatosis in a preterm infant expected to have spontaneous remission.
As a conclusion, ‘Watchful waiting’ may be a treatment option with mesenchymal liver hamartoma in selected very low birth weight preterm infants.