

1. Introduction
Hyper-IgM syndrome (HIGM) features the maintaining or upgrading the level of serum IgM and a slide of IgG, IgE, and IgA levels, with normal numbers of peripheral B cells (1, 2). This immunological phenotype mainly results from the failure of B cells to complete their mutation through immunoglobulin-isotype class switch recombination (CSR) and somatic hypermutation (SHM), which is originated from mutations occurred in CD40, CD40-ligand, activation-induced deaminase, a uracil-DNA glycosylase, and nuclear factor kappa B essential modifier (3).
XHIGM is a rare primary immunodeficiency disorder, and is the most frequent subtype of HIGM (65% - 70%), which is due to mutations in the CD40 ligand molecule expressed transiently on the surface of activated T cells, regarded as a combined T and B immunodeficiency (4, 5).
2. Case Presentation
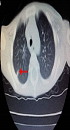
A two-year-old Chinese boy (proband), born to non-consanguineous parents, was diagnosed to be X-link HIGM. He suffered pneumonia and perianal abscess after weaning when he was six months of age. He had recurrent pneumonia, infectious diarrhea, abdominal pain, perianal abscess, anemia, and thrush despite regular intravenous immunoglobulin and anti-infection treatment. He had developed chronic cough one year ago. He got a high fever and pneumonia when admitted to the hospital at his two-years-old. The highest temperature was 40.3°C, with frequent coughing and rapid breathing. On admission, the diagnosis of typical pneumonia was made. His chest CT scan revealed the combination of patch shadow in the right lung and in the back segment of the left lung; the transmittance decreased in the bilateral lower lung, with a dense shadow at the armpit indicating calcification (Figure 1). His parents refused to accept the hematopoietic stem cell transplantation (HSCT) because they could not afford the treatment costs and even refused the common treatment. Finally, he was died at his 2.5-years-old due to serious pneumonia.
, Transthoracic lower dorsal translucency of the lower double lung (B); Reduced double-lower lung permeability, visible patchy patches (C).")
The proband’s chest CT scan. Patches of the left lower lung dorsal patch (A), Transthoracic lower dorsal translucency of the lower double lung (B); Reduced double-lower lung permeability, visible patchy patches (C).
Another patient was the proband’s young brother, who was only one-year-old, suffering from a serious giant perianal abscess at the last admission time. He also had a history of recurrent episodes of abdominal pain, infectious diarrhea, thrush, and perianal abscess since babyhood leading to frequent hospitalization. Because there was not enough money for HSCT after diagnosis, their parents decided to give up treatment and rejected all inspection. Then, the young boy patient died at his 1.2-years-old.
The family history of the two sibling patients was unremarkable and they were born to non-consanguineous parents (Figure 2). The mother’s sisters and brother, even their children, were healthy. There were no similar patients in the history of the family. The grandparents of the sibling boys refused to do any gene test. Therefore, we could not find out the earlier source of the mutation.

Pedigree of three generations of the family with XHIGM syndrome. The two boys got sick, and their mother was the carrier. But we could not know the gene phenotypes of the mother’ sisters, though her children were not sick.
Immunologic evaluation of the two sibling patients was done (Table 1). Both patients had low levels of white blood cell counts, neutrophils, and anemia, but with high levels of stab form granulocyte and blood platelet counts, which may be due to the infection. CD3+ T cells, CD19+ B cells, and CD (16 + 56)+ NK-cell were normal. The level of serum lgG was less than the normal standard, the level of IgA was normal in the proband but less than the normal in the younger brother, and the serum IgM levels in both patients were dramatically over the normal range. The proband had a less detectable expression of CD40L on the surface of peripheral blood lymphocyte, whereas his mother had a drop of CD40L expression compared to a control group made up of healthy test subjects. Nevertheless, the parents refused to do the same test for the second sick boy because of the test cost. By flow cytometry, CD40L expression was 6.7% in the proband, compared to 45.7% in his mother and 83.7% in a healthy control (Figure 3).
| Proband | Young Brother | Reference Value | |
|---|---|---|---|
| Blood text | |||
| White blood cell counts, × 109/L | 2.76 | 1.97 | 4 - 10 |
| Neutrophils, × 109/L | 0.25 | 0.11 | 2 - 7 |
| Lymphocytes, × 109/L | 1.55 | 1.02 | 0.8 - 4 |
| Monocytes, × 109/L | 0.91 | 0.78 | 0.12 - 0.8 |
| Stab form granulocyte, × 109/L | 0.07 | 0.06 | - |
| Hemoglobin, g/L | 56 | 72 | 120 - 160 |
| Blood platelet counts, ×109/L | 506 | 318 | 100 - 300 |
| Lymphocyte counts (%) | |||
| CD3+ | 66.5 | 60.3 | 61.1 - 77.0 |
| CD3+CD4+ | 33.2 | 40.7 | 25.8 - 41.6 |
| CD3+CD8+ | 27.1 | 23.8 | 18.1 - 29.6 |
| CD4+/CD8+ ratio | 1.23 | 1.71 | 0.98 - 1.94 |
| CD19+ | 16.2 | 10.6 | 7.3 - 18.2 |
| CD (16 + 56)+ | 3.8 | 1.96 | 8.1 - 25.6 |
| CD40L (CD154)+ | 0.067 | - | - |
| Immunoglobulins | |||
| IgM, g/L | 7.97 | 9.05 | 0.4 - 2.5 |
| IgG, g/L | 0.35 | 1.96 | 6.0 - 16.0 |
| IgA, g/L | 0.77 | 0.37 | 0.4 - 3.3 |
| sCD40L (ELISA) | 0.12 | 0.17 | 0.53 - 4.23 |
| C-reaction protein mg/mL | 186 | 293 | ≤ 8 |
Immunologic Profile
, compared to 45.68% in his mother (B) and 83.65% in a healthy control (C).")
The CD40L expression on peripheral blood lymphocyte. CD40L expression was 6.74% in the proband (A), compared to 45.68% in his mother (B) and 83.65% in a healthy control (C).
The siblings were also tested for the liver function and renal function, all of which were at the normal levels. Cytomegalovirus (CMV), Epstein-Barr virus (EBV), human immunodeficiency virus (HIV), Pneumocystis jirovecii (PJP), and syphilis were tested negatively. The proband’s chest CT scan revealed the combination of patch shadow in the right lung and in the back segment of the left lung; the transmittance decreased in the bilateral lower lung, with a dense shadow at the armpit indicating calcification.
With the permission of the parents, DNA from both patients and their parents was analyzed to identify the genetic defect. Polymerase chain reaction (PCR) amplification and sequencing of the CD40 ligand (CD40L), AICDA, CD40, and UNG genes were performed as previously described (6). Table 2 mainly presents the primers used for the PCR assays and sequencing. There was no mutation in AICDA, CD40, and UNG genes in the family. A missense mutation (488T>G) in Exon 5 of the CD40L gene was identified in the proband and his brother. This mutation substitutes valine for glycine at codon 163 (V163G) and may produce some undesirous effects. The same heterozygous form (V163G, T>G) was also found in their mother (Table 3 and Figure 4), but no mutation of the CD40L gene in the father. At present, we did not see the same mutation reported yet.
| Name | Sequence | Size of PCR Products |
|---|---|---|
| CD40LG-1-F | AGAGAAGACTACGAAGCACATT | 484 |
| CD40LG-1-R | CAAGATCCTAACCAGAGACAAGA | |
| CD40LG-2-F | AAGTCCTCCTCTTGTTGATGC | 636 |
| CD40LG-2-R | TGTTGACGGCTATTGCTTCC | |
| CD40LG-3-F | GGTTGTTGGCTCATCTGTCA | 535 |
| CD40LG-3-R | GCTGTATGGTTGCTTCTATTAAGG | |
| CD40LG-4-F | TTAGGAGAATGAGACCGATGGAA | 418 |
| CD40LG-4-R | AAGGGAATAGGAGAAGTGTAGGG | |
| CD40LG-5-F | CCTAGATTGGAACATGGCTCTG | 606 |
| CD40LG-5-R | ACCGCTGTGCTGTATTATGAAG | |
| AICDA-1-F | CTGTGTGACTCTGCCTATGACA | 542 |
| AICDA-1-R | CATGGTACAAATCTCAGGACGAA | |
| AICDA-2-F | AATTGTAATCCCAAGTCATCAGCAT | 524 |
| AICDA-2-R | CATCAGCAGGTGGCTCTAAAAGCA | |
| AICDA-3-F | AGTTGCTTAGCCTCTCTGTAACAC | 1601 |
| AICDA-3-R | CCTGCAGTTCTGTTATTCCCAGTAGG | |
| CD40-1-F | CCCCCGATAGGTGGACCGCGATT | 355 |
| CD40-1-R | GAGGCAGCCCACCCCATTCAGC | |
| CD40-2-F | CAAATATCATGCAGGTTCCACT | 304 |
| CD40-2-R | AGTTTACAGTTAACATATGGGGTC | |
| CD40-3-F | TTGAATCCCCTACCCTAAAGCCT | 1215 |
| CD40-3-R | GCAGATGCTTCACAAGTACATGAGT | |
| CD40-4-F | CGTGTTGTGTGCTCAGTGAACCT | 424 |
| CD40-4-R | ACTAAGGAAGTGCTGTTATCATCCCA | |
| CD40-5-F | AAAGACATCTGCCAGCCACATTTCC | 1243 |
| CD40-5-R | TGTTTCTGCATCCAGTGCCAGGTCTC | |
| UNG-1-F | GCGCCAATTGCTGACCGCCACA | 488 |
| UNG-1-R | CCCTCTGATTGGACAGCGCACCGA | |
| UNG-2-F | CTGTTCCACAAATGGGCGTCT | 1099 |
| UNG-2-R | CCAAACCTCGGGATTTCAGTGTT | |
| UNG-3-F | AGTTTAAAATACGCTTAGCCTTTGACC | 573 |
| UNG-3-R | TTCTTAACCAGGGGTGATTTTACGC | |
| UNG-4-F | GGGCTGGCTGTAACTTCTAACCTT | 1083 |
| UNG-4-R | ACAACTCATCACCCCTATAATTTGTCCA | |
| UNG-5-F | TTAAAGTGCAACAGCCACAGTCC | 828 |
| UNG-5-R | TCCCCTGTTTCACCATAAAGGCAAG |
Primers Used for Polymerase Chain Reaction (PCR) Sequencing
| Mutation Site | Gene Mutation | Amino Acid Mutation | Chromosomal Location | Encode Proteins | |
|---|---|---|---|---|---|
| Sibling patients | The coding region of CD40L gene 488th | T-G | V163G | Xq26 | CD40 ligand |
| The mother | The coding region of CD40L gene 488th | Heterozygous mutation, T-G | V163G | Xq26 | CD40 ligand |
| The father | The coding region of CD40L gene 488th | Normal | Xq26 | CD40 ligand |
Patients and Their Family Gene Text Results
 and the mother (B), but not in the father (C). The patient codon 163 changed from GTT to GGT in the patients and their mother. Mutation analysis showed mutations in patients and a heterozygous mutation in the mother. CD40L gene in the father was normal.")
Proband’s family gene sequence results. The CD40L gene mutation at nucleotide position 488 found in the patients (A) and the mother (B), but not in the father (C). The patient codon 163 changed from GTT to GGT in the patients and their mother. Mutation analysis showed mutations in patients and a heterozygous mutation in the mother. CD40L gene in the father was normal.
3. Discussion
We reported the cases of two sibling young boys with the X-linked hyper-IgM syndrome, who were afflicted with repeated infections in the lungs, crissum skin, and the respiratory and intestinal tract. The two patients had the characteristics of HIGM, such as immunodeficiency, normal or high levels of serum IgM, and low levels of serum IgG and IgA (1, 2). The proband had a less detectable expression of CD40L on the surface of peripheral blood lymphocyte, whereas his mother had a drop of CD40L expression compared to a control group made up of healthy test subjects. Mutation analysis revealed the two sibling patients had a missense mutation within Exon 5 of the CD40L gene at nucleotide position 448 (488T>G), which made a valine code (GTT) changing to a glycine code (GGT) at position 168 (V163G) of CD40L protein expression. The same heterozygous form (V163G, T>G) was also found in their mother, but the mother was not sick. Though the parents refused to test for the CD40L expression on the lymphocyte of the young boy due to the cost burden, the mutation of the CD40L gene was enough to demonstrate the disease the boy had. The most important feature was the gene deficiency in CD40L, AICDA, CD40, and UNG genes, but not all the patients can present the mutations. With the gene test, the two sibling boys were found out to have a novel missense mutation in the CD40L gene and their mother was with a heterozygous mutation. It was clear that the mother transferred the mutation gene to the two sibling boys with male disease characteristics of X genetic linkage.
XHIGM is clinically one of the rare immune-deficiencies carried with increasing susceptibility to recurrent severe opportunistic infections, accompanied by low or absent IgG, IgA, and IgE, but normal or elevated IgM levels (1, 2). Nevertheless, with approximately 6.4% low and 42.4% normal IgM serum levels being reported in the literature, the name X-linked “hyper”-IgM syndrome is rather misleading (6, 7). Moreover, X-HIGM patients with normal levels of IgG (8) or IgA (9) have been reported in the literature, as well. Therefore, it may be hard to make a diagnosis and repeated infections and gene mutation testing should be a gold line in the diagnosis.
The most frequent subtype of HIGM (65% - 70%) is an X-linked variant of the hyper-IgM syndrome (XHIGM) stemming from defects in the CD40L gene, which locates at chromosome Xq24 - 27 and encodes a 261 amino-acid protein for the CD40 ligand (CD154) molecule expressed transiently on the surface of activated T cells (4, 5). CD40L is a 39kD glycoprotein that is a member of the TNF family, composed of four structural domains: An N-terminal intracellular tail (amino acids 1 - 22), a transmembrane domain (amino acids 23 - 46), a portion that makes up the extracellular unique domain (amino acids 47 - 122), and the extracellular C-terminal TNF homology (TNFH) domain (amino acids 123 - 261) (10). Defective immunoglobulin class-switch recombination (CSR) and somatic hypermutation (SHM) in HIGM can attribute to the molecular defects in the CD40 ligand/CD40-signaling pathway or to the defects related to the enzymes as the prerequisite of CSR and SHM. Most of the CD40L gene mutations, encoded by Exon 5 and part of Exon 4, are exotic single nucleotide substitutions mainly in the extracellular TNFH domain. Tsai et al. (11) summarized 13 XHIG cases, most of which were mutations at the Exon 5 and the other mutations at the Exon 4. The genetic mutation (488T>G) at the Exon 5 was the observation in our two cases that has never been reported in the past.
Autoimmune diseases are common cases, ranging from thrombocytopenia and seronegative arthritis to inflammatory bowel disease (12). Patients with chronic parvovirus B19 infection 13 also have such manifestations (13). Among all types of HIGM patients, despite the fact that CD27+ isotype-switched memory B cells are notably reduced, patients with a mutation of CD40L do have a normal number of circulating B cells. While central B-cell tolerance relates to B-cell receptor signaling pathways, it is not well characterized for the mechanisms involved in peripheral B-cell tolerance (4, 14, 15). Therefore, we tested the expression of CD40L on the surface of peripheral blood lymphocyte. The CD40L expression in the proband was barely detectable, and the expression in the mother was less than that in the normal person.
HIGM with CD40 and CD40L mutation has roughly fallen into combined T and B immunodeficiency, not predominantly antibody deficiency. 80% of X-linked HIGM patients fail to survive over 25 years of age (1). The chief culprits of high mortality are PJP early and liver disease later in life. It may be beneficial to use trimethoprim-sulfamethoxazole for prophylaxis of PJP and infusions of intravenous immunoglobulin on a monthly basis to help diminish the frequency and severity of infections. Although long-term IVIG treatment will bring IgG levels up, it is less likely to totally suppress IgM levels or prevent all long-term complications such as bronchiectasis and sclerosing cholangitis (16). It was reported that 68% of XHIGM patients were with neutropenia, with 45% of them being chronic. If neutropenia is severe, it may respond to granulocyte colony-stimulating factor (G-CSF) (12). The two boys were severe neutropenia, but their infections were frequent and repeated even when using G-CSF.
The long-term prognosis of XHIGM is guarded. The death can be ascribed to many contributors, covering early-life infections, liver disease, and malignancies. For the reasons, bone marrow transplantation if performed early in life may cure XHIGM, selecting HLA-identical siblings, matched unrelated donors, or partially matched umbilical cord blood (17, 18). Among patients treated with HSCT, treatment at an early age is associated with improved survival (1, 19, 20). It was reported in the literature two XHIGM patients; regular IVIG supplement was ultimately fatal in the first case while the second case underwent hematopoietic stem cell transplantation in the early stage when there was no significant organ damage, as the only cure for XHIGM (11). Gene therapy will be more complicated than for X-linked severe combined immunodeficiency. A dominantly unpleasant response may occur and overexpression may be detrimental, since the expression of the CD40L gene is highly regulated. As a result, strategies for developing gene therapy for the condition will require employing transduction not only of the structural gene but also of the elements for regulating its expression, or adopting methods of utilizing DNA or RNA repair (19, 20).
The parents had the first sick boy when they born the second boy without any prenatal screening for this sickness. If the prenatal screening for this sickness could be done, there should not be the second sick boy. Therefore, prenatal screening is very important for some parents of the kind. As HIGM is a hardly curable disorder, molecular genetic analyses and prenatal diagnoses are expected to possess tremendous value as a screening procedure to identify permutations in HIGM families and to cut the rate of giving birth to HIGM infants.
3.1. Conclusions
In conclusion, if immunodeficiency is diagnosed in a male child, particularly when there is an indistinct death of a male relative, it is suggested taking X-linked diseases as XHIGM into account. It may be hard to make a diagnosis of XHIGM in the normal or low IgM level patients, however, repeated infections and gene mutation testing should be a gold line of the diagnosis. IVIG treatment may be an effective way to make the patients suffer less infection without complete suppressing of IgM levels or to prevent all long-term complications. Bone marrow transplantation and gene therapy should become the best way for XHIGM treatment. Potential families if have mutational analysis may benefit from since it can help disclose carriers and provide appropriate genetic counseling. Upon the diagnosis, prenatal screening may be necessary if the parents want to have the second or more babies.
