








1. Background
Chinese hamster ovary (CHO) cell lines are favorably utilized for the production of recombinant therapeutics protein (1, 2). Metabolic stresses in CHO cell lines usually cause the induction of apoptosis during the production of heterologous protein (3). The induction of apoptosis leads to reduce viability and production of protein (4, 5). It leads to the accumulation of contaminating cellular fragments, diminished culture time, and decreased product and quality (6). Cell death is a significant issue to be dealt with because it affects the viable cell density, product quantity, and quality (7). It is essential to prolong the culture's longevity (viable culture lifespan) to reach a cost-effective level of industrial production (7). It is valuable to block apoptosis in culture to facilitate an extended culture lifetime and increase yield. Because apoptosis is genetically controlled, genetic engineering of key and regulator genes in the apoptosis cascade could be used to prevent or postpone it (6).
Among regulators of apoptosis, caspases are endoproteases that catalyze the breaking of the peptide bond (8, 9). They show a critical function in apoptosis regulation comprising induction, transduction, and amplification of signals; suppressing caspase activation is a beneficial strategy to inhibit apoptosis and increase protein production in the recombinant CHO (rCHO) cell line (7). Caspase-3 plays a fundamental role in apoptosis and is critical to cellular commitment to irreversible apoptotic cell death (10). Inhibition of the caspases-3 expression is one of the approaches for increasing cell viability in the production industry.
Some strategies can be used for genetic engineering to extend bioprocess function and create biologically active and stable proteins (11). The clustered, regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) system opens new avenues in gene editing and manipulation. The most usage of the CRISPR/Cas9 system is the modification and manipulation of DNA. The main advantage of CRISPR technology is the ability to target and cut specific genome regions by specific guide RNA (12). This tool is easy to design, cost-effective, and more efficient than previous genetics of nucleases, including zinc finger nucleases (ZFN) and transcription activator-like effector nuclease (TALEN). It is a simple genome editing system because only one nuclease and a single guide RNA (sgRNA) are used (13). One of the most remarkable things about CRISPR technology is its rapid development. CRISPR/Cas9 system has been applied for CHO genome engineering by knocking out genes correlated to the product yield and quality (14).
Oleuropein is the most noticeable polyphenol component of olives plants (15, 16). It provokes apoptosis by suppressing the PI3K/AKT signaling pathway and activating caspases. A recent study has shown that oleuropein also induces apoptosis via the activation of caspases in HepG2 cell line (17).
2. Objectives
In this study, we used the rCHO cell line expressing human erythropoietin for manipulating the caspase-3 gene. By targeting the caspase-3 gene using the CRISPR/Cas9 system, we tried permanently to inhibit the expression of the caspase-3 protein (Figure 1). Decreasing caspase-3 expression can inhibit apoptosis and increase cell viability and density. We induced apoptosis by using different concentrations of oleuropein in manipulated CHO cell lines. Finally, the amount of erythropoietin produced in the presence and absence of oleuropein was measured at the manipulated and control cell lines.

A schematic illustration of gene manipulation for increasing the yield of the cell line.
3. Methods
3.1. In Silico Analysis
Information relevant to the caspase-3 gene was conveyed to the STRING server (https://string-db.org/) to generate the gene network. Knockout of the target gene and its pathway were checked. The protein-protein interaction network was drawn, and the effect of the caspase-3 gene on the apoptosis pathway and disrupts this network was observed.
3.2. Plasmid Construction and gRNA Design
Plasmid pLenti-U6-sgRNA-SFFV-Cas9-2A-Puro contains U6 and SFFV promotor, sgRNA, Cas9, 3′LTR, and 5′LTR plasmid was purchased from an abmgood company (abmgood, Canada). sgRNAs were designed against the caspase-3 gene in rCHO by the online tool Optimized CRISPR Design (http://crispr.mit.edu/), and the sgRNAs with rarer off-target positions were selected for further investigation (18, 19). sgRNA sequences are listed in Table 1.
| No. | Sequence | Direction | Mismatch | Position |
|---|---|---|---|---|
| 1 | Target 1: GTC-GGT-AAG-AAC-GGC-ACA-GC | 3′→5′ | 0 | 6572-6591 |
| 2 | Target 2: ACT-AGG-GAC-CGA-CTT-TAT-CG | 5′→3′ | 0 | 1247-1266 |
| 3 | Target 3: GAT-TTT-CCT-TAC-AAC-CGT-GG | 3′→5′ | 0 | 4037-4056 |
The Sequence of sgRNAs
3.3. Cell Culture and Transfection
Recombinant CHO adherent cells producing erythropoietin were obtained from the production and research complex of the Pasteur Institute of Iran. The rCHO was cultured in DMEM/F-12 medium in T-25 cm2 vented cap tissue culture flasks, supplemented with 10% FBS, and incubated in a humidified chamber at 37°C with 5% CO2. A total of 1×103 cells were transfected with the designed plasmid. Transfections were carried out by ScreenFect A plus (Wako, Japan) according to the manufacturer's instructions. After successful transfection and antibiotic selection, Puromycin–resistant cells detached by trypsin, and the single clones were isolated by serial dilution in DMEM/F-12 (20% FBS).
3.4. DNA Extraction
The cells were harvested and pelleted by centrifugation in a 1.5 mL tube and resuspended on 100 µl PBS buffer. Then, Genomic DNA was extracted by DNG-PlusTM reagent according to the manufacturer's instructions (Sinaclon, Iran).
3.5. PCR Amplifications and Sequencing
PCR amplification was done by master mix (Thermo Scientific, USA) and specific caspase-3 primers. PCR products were electrophoresed in agarose gel and visualized by DNA safe stain. The purified PCR products were sequenced 3130XL genetic analyzer (Applied Biosystem, USA).
3.6. Real-time PCR
Whole RNA was obtained from the cell pellets using RNX-plusTM solution (Sinaclon, Iran). Complementary DNA (cDNA) was synthesized from 700 ng of extracted RNA with Moloney murine leukemia virus reverse transcriptase and random hexamer primers by the Viva cDNA kit (Vivantis, Malaysia). Gene expression levels of caspase-3 were evaluated and normalized against GAPDH genes. The 2-ΔΔCt method was used to calculate the relative fold gene expression.
3.7. Cell Proliferation Assay
The manipulated and control cell lines were cultured at 0.5 × 106 densities. The cell density was determined by counting and using an Improved Neubauer haemocytometer (20, 21), and viable cells were distinguished from dead cells by the Trypan blue dye exclusion method.
3.8. Induction of Apoptosis and MTT Test
Around 5,000 cells from the manipulated and control cell lines were seeded into 96-well plates in DMEM/F-12 in triplicate. After overnight incubation, the medium from each well was removed and replaced with a medium containing different concentrations (500, 1,000, 2,000, 4,000, and 8,000 µM/mL) of oleuropein (Sigma-Aldrich, USA) for 24 h and 48 h. All cell lines were incubated in a 0.5 mg/mL MTT reagent at 37°C for four hours. Then, the medium was discarded, and purple formazan crystals were dissolved by adding 150 µl isopropanol per well. In the end, the absorbance of the colored solution was recorded at 570 nm wavelength by a microplate reader (22, 23).
3.9. Western Blot
Manipulated and unmanipulated rCHO cells were lyzed and loaded on an SDS-PAGE gel (12%) and transferred to the PVDF membrane (Bio-Rad, USA). Then, they were incubated with anti-caspase 3 (Cat No: ab184787, Abcam, USA) and anti-beta actin antibodies (Cat No: ab8227; Abcam, USA) for one hour at RT. Successively, membranes were washed three times and incubated to buffer, including goat anti-rabbit IgG H&L (HRP). The expression of proteins was normalized by β-actin. Densitometry of bands was done by the Gel Analyzer Version 2010a software (NIH, USA)
Measurement of Erythropoietin: Manipulated and unmanipulated rCHO cell lines producing recombinant human erythropoietin (EPO) were grown in T-flasks. After one day of culture, the FBS medium was replaced with a serum-free production medium (with and without 2,000 µM oleuropein). The concentration of EPO secreted into the culture media was assessed by an EPO ELISA Kit (antibodies-online, Germany) according to the manufacturer's guidelines at T = 0, T = 24, T = 48, T = 72, and T = 96.
3.10. Statistical Analysis
Statistical analysis was done by GraphPad Prism software (GraphPad Software). Comparisons of caspase-3 gene expression, viability, and EPO production between treated and untreated cell lines were evaluated by the Student's t-test (two groups) or a t-test analysis of variance (ANOVA).
4. Results
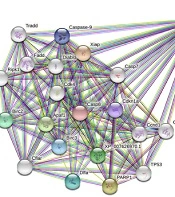
The Caspase-3 is well-known as an executioner caspase in the apoptosis pathway. By the STRING database, the Caspase-3 apoptotic regulating gene network of the Chinese hamster (Cricetulus griseus) was drawn up. There was a minimum interaction score of 70% (high confidence) and a maximum of 50 genes that could interact (Figure 2).

The result of STRING analysis of caspase-3 in Cricetulus griseus.
After successful transfection and antibiotic selection, single clones were isolated by serial dilution in DMEM/F-12 (20% FBS). Ninety individual clones were picked up and sequenced by the dideoxy terminal method. All the DNA sequences were aligned with control by Clustal online tool (http://www.ebi.ac.uk/Tools/msa/clustalo/). The site of cleavage and sequence alignment of control and some sequence-manipulated clones are shown in Figure 3.
 PCR product sequencing of the control cell line by Sanger sequencing, and (B) edited cell line (clone-62) containing AATC insertion, (C) Different individual single-cell PCR product alignments. Clone 62 showed the insertion of four nucleotides after breaking repair. (D) viable cell density. Comparison between the manipulated CHO cells (clone-62) and the control ones after 72 h. Results represent the mean of three analyses, and error bars show the standard deviation (P-value < 0.0017). (E) Expression of caspase-3 gene in manipulated cell lines (clone-62) compared to the control ones.")
(A) PCR product sequencing of the control cell line by Sanger sequencing, and (B) edited cell line (clone-62) containing AATC insertion, (C) Different individual single-cell PCR product alignments. Clone 62 showed the insertion of four nucleotides after breaking repair. (D) viable cell density. Comparison between the manipulated CHO cells (clone-62) and the control ones after 72 h. Results represent the mean of three analyses, and error bars show the standard deviation (P-value < 0.0017). (E) Expression of caspase-3 gene in manipulated cell lines (clone-62) compared to the control ones.
For evaluating cell growth and density, 500,000 cells were seeded in a T-25 flask and counted after 72 h. Figure 3 shows viable cell density between the control cells and the manipulated cell lines (clone-62). Significant differences in the viable cell density were observed between the two groups (the control and clone-62) (142%, P-value < 0.0017).
RT-PCR with sequence-specific primers confirmed that caspase-3 expression was decreased 2.7-fold at the mRNA level in the edited cell line (clone-62) compared to the control cell line (P-value < 0.0005) (Figure 3E).
The Western blot method confirmed the reduced expressions of caspase-3 by the CRISPR/Cas9 system (Figure 4). β-actin was used as an internal control for normalizing protein expression. Western blot analysis of clone-62 showed a more than 6-fold reduction in the caspase-3 expression gene compared to the control (P-value < 0.0001).
 Caspase-3 protein decreased in clone-62 (individual manipulated cell) compared to control. (B) The expression level of caspase-3 protein in manipulated cell (clone-62) by CRISPR/Cas9 system has decreased 6.4-fold compared to the control.")
Evaluation of caspase-3 protein expression by western blot. (A) Caspase-3 protein decreased in clone-62 (individual manipulated cell) compared to control. (B) The expression level of caspase-3 protein in manipulated cell (clone-62) by CRISPR/Cas9 system has decreased 6.4-fold compared to the control.
The viability of the manipulated clone cells (clone-62) and the control ones was evaluated in the presence of different concentrations of apoptosis inducers (500, 1,000, 2,000, 4,000, and 8,000 µM/mL oleuropein) after 24 h and 48 h incubation (Figure 5A-B). As shown in figure 5, the manipulated cell exhibited resistance to apoptosis in the presence of oleuropein after 24 (A) and 48 (B) hours of incubation and confirmed by MTT assay.
For evaluation of the resistance to apoptosis of the edited cell line, the manipulated and control cell lines were treated with 500, 1,000, 2,000, 4,000, and 8,000 µM/mL of oleuropein for 24 and 48 hours, and IC50 was calculated (Figure 5C-D). The IC50 of manipulated cells was higher than control cells (7271 µM/mL Vs. 5741 µM/mL) after 48 h.
To investigate the knockdown of caspase-3 on the expression of EPO, the amount of erythropoietin produced in the manipulated cell line (Clone-62) and the control cell line was measured by the ELISA method. The results showed production of EPO in the manipulated cell line (clone-62) is higher than in the control ones (Figure 5). This increase in the production of EPO is significantly higher in clone-62 in the presence of 2,000 µM oleuropein compared to the control one after 96 hours.
 and control. The viability was measured in the presence of different concentrations of apoptosis inducers, oleuropein, after 24 h (A) and 48 h incubation (B). Comparison of the IC50 value in the presence of 500, 1,000, 2,000, 4,000, and 8,000 µM/mL of oleuropein. (C) IC50 value of control and manipulate cell line (clone-62) at T = 24 h, and (D) IC50 value of control and manipulate cell line (clone-62) at T = 48 h. Erythropoietin production, Control cell line, and clone-62 (manipulated cell) in the medium without oleuropein (E), and in the presence of 2,000 µM/mL oleuropein (F).")
Cell viability assessment of manipulated cell lines (clone-62) and control. The viability was measured in the presence of different concentrations of apoptosis inducers, oleuropein, after 24 h (A) and 48 h incubation (B). Comparison of the IC50 value in the presence of 500, 1,000, 2,000, 4,000, and 8,000 µM/mL of oleuropein. (C) IC50 value of control and manipulate cell line (clone-62) at T = 24 h, and (D) IC50 value of control and manipulate cell line (clone-62) at T = 48 h. Erythropoietin production, Control cell line, and clone-62 (manipulated cell) in the medium without oleuropein (E), and in the presence of 2,000 µM/mL oleuropein (F).
5. Discussion
Growing request for therapeutic proteins obligates the development of cost-effective technologies and policies for high-quality and quantity expression systems in the biopharmaceutical industry (11). CHO cell lines are generally used as the mammalian host because of their safety for human usage (12). CHO cells must be propagated in big bioreactors at high density and concentrations to respond to the market's demands in large-scale production. Hence, improving this kind of expression system is of great commercial benefit (24).
The byproducts (lactate and ammonia) accumulate during the cell cultures in bioreactors. They have led to the induction of apoptosis and declined cell viability and density in bioreactors. Several approaches, such as nutrient feeding, anti-apoptotic chemicals, and genetic engineering, have been applied to prolong cell viability (25). The engineering of anti-apoptosis has been one of the major research fields in cell line improvement for protein production. So, it is of meaningful value to increase the concentration of viable cells by down-regulating or preventing apoptosis in culture via suppressing cell death, expanding culture lifetime, and boosting cell-specific productivity by holding cellular activity (26, 27). Overexpression of some anti-apoptotic genes (Mcl-1, 30Kc6, Bcl-2, Bcl-w, Aven, and E1B-19K) and down-regulation of pro-apoptosis genes expression (Bax, Bak, and Bok) in mammalian cells have been studied to increase recombinant protein production (27). Zhang et al. reported an 82% increase in the production of antibodies in CHO cells co-transfected with Bcl-xl and a 34% increase by co-transfected with Mcl-1 (28).
Different anti-apoptotic plans have been examined. For example, many efforts have been made by gene knockdown using small interfering RNAs (siRNAs) (4) and microRNAs (mRNAs). However, significant problems such as off-targeting, temporary inactivation of gene function (29), and generation of gene knockouts prohibited these methods (30). The silencing effect was relatively unstable in many of the isolated cell lines. Shen et al. showed that siRNA inhibition confers a 32% reduction of PDK1 mRNA level, and it needs multiple selection rounds for the high degree of gene knockdown (31). Many knockdown lines lost their RNAi effect on gene expression during routine subculture (4), and a high level of gene knockdown may require several selection steps (31). Moreover, the necessity to present a reporter gene (GFP or luciferase) is not a favorite for developing cell lines used in the industry (32). For the first time, Han and Rhee have shown that exosomes derived from CHO cells can decrease apoptosis during the cell culture when supplemented with the culture medium (33). However, this method requires additional work and materials. Cost et al. showed complete and permanent Bax and Bak protein elimination by ZFN in CHO without affecting cell growth (34). Figueroa et al. demonstrated that the expression of anti-apoptosis protein increased viable cell concentration, but the engineered cells showed a reduction in growth rate (35).
The caspase knockdown trials in CHO cells generally led to minor improvements in viable cell concentration or viability of up to 40% (36). Apoptosis resistance had been presented by gene inhibition of Casp-3 and Casp-7 to overcome apoptosis in CHO cells (by CRISPRi) (3). Some inhibitors of specific caspase have been assessed, but in large-scale cultures might make them unaffordable because of the cost. Quieting of caspase-3 utilizing the anti-sense expertise is another strategy for inhibiting apoptosis, significantly suppressing apoptosis, and extending the culture lifespan. However, this method did not translate into improved volumetric yield because of the loss of metabolic capacity due to changes in the membrane of the mitochondria (10). The advantage of caspase-3 inhibition is that paths of apoptosis signaling converging on some important executioner genes can be hindered concurrently (25).
Editing the genome by CRISPR/Cas9 is helpful for establishing genome-engineered cells (24, 30). It has become a progressively significant view of cell line manipulation for developing the production of recombinant proteins (13). In comparison to other nucleases, zinc finger nucleases (ZFN), and transcription activator‐like effector nucleases (TALEN), the CRISPR/Cas9 system is significantly more accessible and more specific (29, 37, 38). Furthermore, a recent study showed that CRISPR/Cas tool has far fewer off-target than RNAi (39).
The variety, flexibility, effectiveness, and simplicity of CRISPR systems that may be applied have indeed developed cell engineering (12) and the ability to address many target sites with multiple gRNAs simultaneously (40). It is an attractive tool for genome editing for academic and industrial groups (41). The CRISPR/Cas system is more flexible than ZFNs and TALEN, which apply RNA-based DNA targeting (42). Some studies have shown the excellent function of CRISPR/Cas9 in CHO with knockouts or insertion of genes, targeting to influence the product quantity and quality of yields (32). Xiong et al. found that CRISPRi can successfully repress Bak and Bax genes and rescue CHO cells from apoptosis (3). Shen et al. enhanced recombinant protein production by CRISPRi in CHO cells without impeding cell growth (14).
The CRISPR/Cas9 method is precise in principle, but in reality, not so much. It can make mutations elsewhere in the genome, known as “off-target” variation. Various online programs have been created and effectively applied to apply and guess off-target attachment in silico (43) and minimize the possibility of off-target genome disruption (44). Third-party alignment tools are introduced to find off-target sites such as BWA and Bowtie (45). Shortening the length of gRNA to less than 20 bases has a significant effect in reducing off-targe. Fu et al. showed that truncated gRNAs could reduce some off-target by 5000-fold without affecting on-target genome editing efficiencies (46). Of course, gRNA with less than 15 nucleotides are not safe because they have loss-of-function and specificity (47). In addition to the off-target, other consideration such as polymorphism, delivery method, and ethics should also be considered.
In this project, we display the effective manipulation of caspase-3 by CRISPR/Cas9 to create a modified rCHO cell line on a laboratory scale with long viability. These cells are more resistant to adverse environmental conditions. Their IC50 was higher than control cells and also showed more cell proliferation in the presence of apoptotic inducers. We also used the scratch wound healing proliferation assay to detect cell proliferation in the presence of oleuropein (a stimulant of apoptosis) in high-density conditions (data not shown). Results showed that cell density was significantly increased by manipulating the caspase-3 gene even in the presence of apoptotic stimuli (142%, P-value = 0.0017). The high-level production of recombinant proteins is directly correlated with a high density of cells. The density of cells is closely tied to cell environmental conditions. By manipulating apoptotic genes in unfavorable conditions and the presence of an apoptotic inducer, recombinant protein-producing cell lines can produce more culture yield. This suggests that the inhibition of apoptosis in the manipulated cells allowed them to keep their activity of cells for an extended time to permit cell growth and production of EPO protein. Our results display that CRISPR/Cas9 system is a suitable tool for rCHO cell engineering and proposes a substitute system for ZFN, TALEN, and siRNA. These data collectively warrant the potential of CRISPR/Cas9 for CHO cell engineering and lead to developed culture viability and host yields.
5.1. Conclusions
The cost-effective production of recombinant proteins in the industry is one of the Health system's priorities. However, the production of recombinant proteins on an industrial scale is costly. As a result, we need to optimize production to produce them cost-effectively. Manipulation of the host cell genome is one of the most effective ways in this regard. There is less need to add expensive substances and molecules in the production process to prevent apoptosis in these methods. In addition, the CRISPR/Cas9 method produces stable cell lines. Many studies have shown that stable cells can produce recombinant proteins by reducing the expression of apoptosis-inducing factors. Extended culture lifetime and viability can be translated into developing recombinant protein quantity and quality. These cells produce the recombinant proteins with more intensity and at a longer time, reducing the product's cost.
