




1. Background
2. Objectives
3. Methods
3.1. Clinical Samples
3.2. Cell Culture, Establishment of DDP-Resistant Cells, and Treatments
3.3. Cell Transfection
3.4. Quantitative Real time Polymerase Chain Reaction (RT-qPCR)
3.5. Western Blotting
3.6. Cell Counting Kit-8 (CCK-8)
3.7. Colony Formation
3.8. Detection of MDA, GSH, Fe2+, ROS Levels
3.9. Animal Model
3.10. Immunohistochemical Staining
3.11. Statistical Analysis
4. Results
4.1. DHCR24 Was Highly Expressed During DDP Resistance
 analysis of DHCR24 mRNA level in DDP-resistant and sensitive NSCLC tissues and para-carcinoma tissues; B and C, DHCR24 mRNA and protein expression in DDP-resistant NSCLC cells and parental cells was assessed by RT-qPCR and Western blotting; D, GEPIA database analyzed the correlation between DHCR24 expression and survival of NSCLC patients. *** P < 0.001.")
The DHCR24 was up-regulated in DDP-resistant NSCLC samples and cells. A, quantitative real time polymerase chain reaction (RT-qPCR) analysis of DHCR24 mRNA level in DDP-resistant and sensitive NSCLC tissues and para-carcinoma tissues; B and C, DHCR24 mRNA and protein expression in DDP-resistant NSCLC cells and parental cells was assessed by RT-qPCR and Western blotting; D, GEPIA database analyzed the correlation between DHCR24 expression and survival of NSCLC patients. *** P < 0.001.
4.2. DHCR24 Down-Regulation Raised DDP Sensitivity
 and Western blotting determined DHCR24 mRNA and protein levels; C and D, after exposure to various concentrations of DDP, cell viability was assessed by CCK-8; E-G, the growth of NSCLC cells was evaluated by colony formation assay. * P < 0.05, *** P < 0.001.")
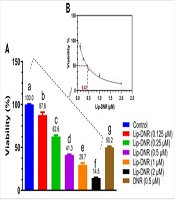
The DDP sensitivity was elevated in DHCR24-silenced NSCLC cells. A549/DDP and NCI-H1975/DDP cells were transfected with sh-DHCR24 or sh-NC. A and B, quantitative real time polymerase chain reaction (RT-qPCR) and Western blotting determined DHCR24 mRNA and protein levels; C and D, after exposure to various concentrations of DDP, cell viability was assessed by CCK-8; E-G, the growth of NSCLC cells was evaluated by colony formation assay. * P < 0.05, *** P < 0.001.
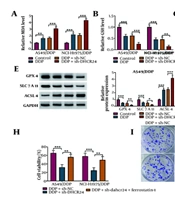
4.3. DHCR24 Deficiency Induced Ferroptosis to Enhance DDP Sensitivity of NSCLC Cells

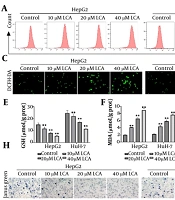
The DHCR24 knockdown enhanced DDP sensitivity of NSCLC cells via inducing ferroptosis. Sh-DHCR24 or Sh-NC-transfected A549/DDP and NCI-H1975/DDP cells were treated with 10 μg/mL DDP. A-D, the levels of MDA, GSH, Fe2+, and ROS levels were detected by commercial kits. E and F, the protein abundance of GPX4, LC7A11, and ACSL4 was measured by Western blotting. Sh-DHCR24-transfected A549/DDP and NCI-H1975/DDP cells were treated with 10 μM Ferrostatin-1 in the presence of DDP; G, cell viability of DDP-resistant NSCLC cells was evaluated by CCK-8; H, the growth ability was analyzed by colony formation assay. * P < 0.05, ** P < 0.01, *** P < 0.001.
4.4. DHCR24 Knockdown Triggered Ferroptosis via Inactivation of PI3K/AKT/GSK3β Pathway

The DHCR24 down-regulation triggered ferroptosis by inactivating PI3K/AKT/GSK3β pathway. A549/DDP and NCI-H1975/DDP cells were transfected with sh-DHCR24 or Sh-NC together with treatment with 10 μg/mL DDP. A and B, the protein levels of p-PI3K, PI3K, p-AKT, AKT, p-GSK3β, and GSK3β were detected by Western blotting; C-F, MDA, GSH, Fe2+, and ROS levels were assessed by commercial kits. Sh-DHCR24-transfected DDP-resistant NSCLC cells were co-treated with 100 ng/mL IGF-1 upon DDP exposure. G and H, the protein abundance of GPX4, LC7A11, and ACSL4 was determined by Western blotting; I and J, the growth of cells was determined by CCK-8 and colony formation assay. * P < 0.05, ** P < 0.01, *** P < 0.001.
4.5. DHCR24 Depletion Restrained Xenograft Growth and DDP Resistance in Vivo
; D, Western blotting analysis of GPX4, LC7A11, ACSL4, p-PI3K, PI3K, p-AKT, AKT, p-GSK3β, and GSK3β protein levels in tumor tissues. * P < 0.05, ** P < 0.01, *** P < 0.001.")
The DHCR24 silencing delayed xenograft growth and enhanced DDP sensitivity of NSCLC cells in vivo. The A, tumor volume; and B, weight were detected in different groups; C, Ki67 and DHCR24 expression in tumors was determined by immunohistochemical staining (magnification, x400); D, Western blotting analysis of GPX4, LC7A11, ACSL4, p-PI3K, PI3K, p-AKT, AKT, p-GSK3β, and GSK3β protein levels in tumor tissues. * P < 0.05, ** P < 0.01, *** P < 0.001.