The present study combines the rich knowledge of TCM with modern computational methods to identify potential drug candidates and develop novel therapeutic agents for breast cancer treatment (
10). Many TCM formulations and herbs have been used for centuries to treat various ailments, including cancer. Today, with the advent of modern computational techniques, researchers are analyzing compounds from these plants and understanding their probable mechanisms for treatment in breast cancer (
11). Computer-aided drug designing studies generally use virtual screening and molecular docking techniques to predict active molecules from the vast Chinese medicine database that can bind with selected targets of breast cancer. Simulating the binding of TCM compounds to target proteins allows researchers to identify lead-like natural pseudo-ligands. Typically, Traditional Chinese Medicine treats health and disease in terms of whole system affinities rather than a single-target approach (
12). However, molecular docking and virtual screening have their limitations. Relying completely on static protein structures is one of the setbacks of molecular docking, as it may not accurately represent biological conditions. Solvation effects and protein flexibility are often neglected, and scoring functions can be inaccurate. Virtual screening methods may also fail to fully interpret the complex interactions occurring in a cellular environment. Moreover, molecular docking cannot assure bioactivity without subsequent experimental validation.
Computer-aided drug designing studies are conducted to identify potential TCM compounds and formulate them for targeting multiple breast cancer targets, which may lead to superior therapeutic efficacy (
13). Computer-aided drug designing has significant potential to discover and optimize potent bioactive molecules from Traditional Chinese Medicine for their therapeutic applications in breast cancer treatment (
4). The results of this study and the analysis highlight the binding affinities of these ligands against key proteins involved in cancer pathways: Epidermal growth factor receptor (EGFR, PDB ID: 1XKK), aromatase (PDB ID: 3S7S), and phosphoinositide 3-kinase alpha (PI3K alpha, PDB ID: 7PG6). In the present study, the docking results for dipiperitylmagnolol across three different proteins (1XKK, 3S7S, and 7PG6) consistently show stronger binding affinity compared to control ligands (lapatinib, exemestane, and alpelisib). The stronger MolDock scores, interaction energies, and total scores for dipiperitylmagnolol indicate that it forms more stable and favorable interactions with these proteins. In contrast, while control ligands demonstrate good binding, they are generally outperformed in terms of overall binding strength.
Dipiperitylmagnolol's higher binding affinity may offer potential therapeutic advantages, as stronger overall interactions can contribute to more effective inhibition of the target proteins. Similarly, Dzobo employed several computational approaches, including molecular docking, molecular dynamics simulations, ADMET, and DFT, to identify and examine potential leads from TCM sources with prospective anti-breast cancer activities (
8). Shimu also applied various computational methods, such as molecular docking, molecular dynamics simulations, and ADMET analysis, to evaluate TCM-derived compounds with potential anti-breast cancer activity. Their study focused on identifying lead compounds from TCM sources through virtual screening and molecular docking, similar to the approach used in this study (
14).
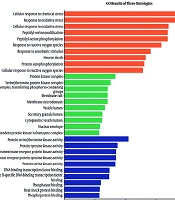
Moreover, the potential of plant-derived compounds in cancer treatment has been widely recognized. This is due to the ability of these natural compounds to interfere with cancer progression through various mechanisms, including apoptosis, angiogenesis, and the inhibition of cancer-related enzymes such as aromatase and PI3K (
15). Dipiperitylmagnolol's higher binding affinity may offer potential therapeutic advantages, as stronger overall interactions can contribute to more effective inhibition of the target proteins. Similarly, Dzobo employed several computational approaches, including molecular docking, molecular dynamics simulations, ADMET, and DFT, to identify and examine potential leads from TCM sources with prospective anti-breast cancer activities (
8). Shimu also applied various computational methods, such as molecular docking, molecular dynamics simulations, and ADMET analysis, to evaluate TCM-derived compounds with potential anti-breast cancer activity. Their study focused on identifying lead compounds from TCM sources through virtual screening and molecular docking, similar to the approach used in this study (
14). Moreover, the potential of plant-derived compounds in cancer treatment has been widely recognized. This is due to the ability of these natural compounds to interfere with cancer progression through various mechanisms, including apoptosis, angiogenesis, and the inhibition of cancer-related enzymes such as aromatase and PI3K (
15). Plants have long been recognized as a rich source of bioactive components responsible for various medicinal properties (
16). In the present study, we examined numerous plant-derived compounds and their anti-aging benefits. Given that thousands of bioactive compounds are present in different plants, the potential for developing new pharmaceuticals and natural remedies is immense (
17). These findings pave the way for future research exploring the therapeutic potential of these compounds in pharmaceuticals and healthcare (
18). The present study highlights the importance of plant-derived bioactive compounds and their medicinal uses, which can further drive additional research in the fields of natural medicine and drug discovery. Out of the 129 herbal compounds analyzed, dipiperitylmagnolol demonstrated superior performance at multiple binding sites of three different protein targets of breast cancer. As a result, dipiperitylmagnolol was identified as the most promising candidate for multiple protein binding sites (1XKK, 3S7S, and 7PG6), making it a strong drug candidate for further investigation in breast cancer therapy (
19). Additionally, sophoradochromene and sophoranone exhibited robust binding affinities at specific protein binding sites. The study also identified a range of compounds with potential therapeutic applications for breast cancer, including sophoradochromene, sophoranone, and arctigenin, which consistently showed favorable docking scores across multiple sites. This information can guide researchers in selecting ligands for specific binding sites to facilitate structure-based design of targeted drugs or treatments. Notably, the study revealed that
Magnolia species, including
M. officinalis, are significant sources of bioactive compounds with high affinities toward breast cancer proteins, underscoring their therapeutic potential (
20). Thought to exert both anti-inflammatory and antiviral activity,
Magnolia species have been widely used in TCM. Indeed, it is a folk remedy for many conditions, including cancer, which presumably reflects its anticancer potential. Compounds derived from
Magnolia species have been demonstrated to modulate many of the same signaling pathways involved in cancer progression, making them compelling leads for further investigation (
21).
Magnolia species have been identified as the most common plant sources for many compounds in our dataset, likely indicating that these plants are a rich source of bioactive molecules with potential therapeutic implications for breast cancer. Furthermore, it is worth emphasizing the importance of screening different plants to discover new therapeutic agents, as other species belonging to the Magnolia genus may yield specific bioactive compounds.
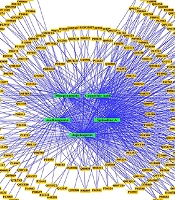
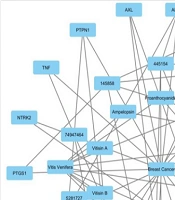
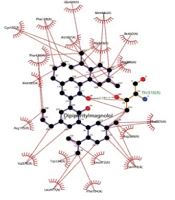
The provided data could facilitate the selective isolation and production of bioactive compounds from these plant resources, potentially contributing to the development of new breast cancer treatments. Another important aspect of this research is the construction of interaction networks. These networks illustrate the connectivity between bioactive compounds, their plant sources, and target proteins, providing a comprehensive view of the TCM pharmacopoeia (
22). The construction of these interaction networks highlights the links among top-hit compounds, plant sources, and target proteins. Understanding these intricate networks offers a structural basis for the action of compounds on proteins, identifying essential proteins and their associated plant sources, and presenting a complete dataset perspective.
These networks can be used by researchers to explore potential synergistic effects or combinatorial treatments between different compounds and plant sources. Additionally, the data from the ADME-Toxicity study were crucial for understanding the pharmacokinetic properties and potential safety hazards of these compounds. This information provided a foundation for prioritization in preclinical rodent toxicity-testing programs and informed subsequent clinical development strategies for selected breast cancer therapeutics (
23).
The ADME-Toxicity analysis in this study demonstrated that the top docking hits for the three target proteins fall within the acceptable limits for FDA-approved drugs, underscoring their potential as promising drug candidates. Dipiperitylmagnolol, in particular, exhibits good bioavailability and a low toxicity profile. It has a QED of 0.312, indicating moderate drug-likeness. Although this value is below the optimal threshold of 0.5, its bioavailability of 0.65 exceeds the 0.55 threshold, suggesting adequate absorption. This bioavailability, combined with its low clinical toxicity (0.03), positions dipiperitylmagnolol as a highly interesting candidate for further investigation. The toxicity analysis of dipiperitylmagnolol reveals that it has a unique safety and pharmacokinetic profile, enhancing its potential as a drug candidate. Dipiperitylmagnolol demonstrates a longer half-life and exhibits good caco-2 permeability (-5.41) along with excellent hydration properties (-3.48), indicating favorable intestinal absorption and solubility. Understanding these ADME properties and associated toxicological risks is crucial for advancing these molecules in drug discovery.
A systematic application of this methodology provides a comprehensive assessment of compound effectiveness and toxicity, which is critical for the bench-to-bedside translation of potential drugs. While CADD studies show great promise for the application of TCM in breast cancer therapy, several challenges need to be addressed before these methods can be fully embraced. These include rigorous experimental validation and standardization of the formulations used (
24). In addition, these results underscore the relatively limited understanding of the bioavailability and pharmacokinetic nature of TCM compounds, which must be further explored to facilitate their clinical translation. The DFT analysis highlights the molecular regions where the HOMO and LUMO exhibit significant electron density, offering insights into their electronic properties. However, experimental validation remains a key area requiring greater focus, beyond merely validating control statistics and establishing quality standards, which are essential steps for making TCM-based drugs credible and reliable. While CADD studies effectively identify candidate lead molecules, their translation to clinical use necessitates thorough pharmacokinetics and pharmacodynamics analyses. The ultimate steps in validating TCM-based breast cancer therapies involve comprehensive safety and efficacy assessments through clinical trials. Such a holistic approach will support broader efforts to identify new treatments for breast cancer, reinforcing the potential of plant-derived compounds and paving the way for further investigations. The combinatorial use of herbal therapies with conventional treatments holds promise for enhancing therapeutic efficacy in breast cancer treatment. These findings provide a foundation for developing innovative treatments that could offer hope to patients and their families affected by breast cancer. However, certain limitations of this study must be acknowledged. The reliance on network pharmacology may oversimplify the complex biological interactions involved in breast cancer. Additionally, molecular docking results, while insightful, may not fully capture in vivo conditions or provide an accurate reflection of physiological responses. Furthermore, the lack of experimental studies to confirm the efficacy of the herbal compounds presents a significant gap, highlighting the need for more robust experimental validation to bridge the gap between computational predictions and clinical applicability.
4.1. Conclusions
This study concludes that dipiperitylmagnolol is a potent anti-cancer candidate for breast cancer therapy. The docking studies demonstrated the most favorable docking scores and binding affinities across three targeted breast cancer proteins. Magnolia species were identified as significant sources of bioactive compounds with substantial therapeutic potential.
A combinatorial approach that integrates herbal therapies with conventional treatments could enhance therapeutic efficacy by leveraging unique mechanisms of action to improve patient outcomes. This synergy may reduce side effects, enhance drug absorption, and address the challenge of disease resistance, offering a novel strategy for breast cancer management.
Therefore, the study strongly recommends the exploration and use of herbal compounds for targeted breast cancer treatment.


 at the active site of 1XKK, 3S7S and 7PG6. The docking results showed the binding affinities between dipiperitylmagnolol and the protein active sites (1XKK, 3S7S and 7PG6), highlighting key interactions such as hydrogen bonding, hydrophobic contacts, and van der Waals interactions.")

 analysis of 1XKK-dipiperitylmagnolol; B, 3S7S-dipiperitylmagnolol; and C, 7PG6-dipiperitylmagnolol during the 100 ns MD production. The graph shows a stable RMSD which indicates that dipiperitylmagnolol is well-bound to the active site of the proteins (1XKK, 3S7S and 7PG6) = maintaining its conformation throughout the simulation, implying a potential stable inhibitor.")
 in the ligand throughout the simulation trajectory from 0 ns to 100 ns.")
 plot of 1XKK-dipiperitylmagnolol; B, 3S7S-dipiperitylmagnolol; and C, 7PG6-dipiperitylmagnolol during the 100 ns MD production. The plot provides insights into the correlation between protein-ligand interaction components and its binding free energy.")
 carcinogenicity vs acute toxicity LD50 of the top 5 docking hits of each of the target proteins. (Dipiperitylmagnolol, sophoranone, arctigenin, -(S)-rosmarinic acid, sophoradochromene, gingerenone A, gamma-tocotrienol, piperitylhonokiol, Licoagroisoflavone, phaseol and gingerenone B)")