Laboratory-grade solvents, pentylenetetrazol (PTZ), potassium carbonate, and all reagents were purchased from Merck (Merck, Germany). 1-Methyl-1H-indazole-3-carbonyl chloride (CAS No: 106649-02-9) was obtained from Pharmaffilates (Parchkula, India). A Bruker 500 MHz Ultra spectrometer (Bruker, Darmstadt, Germany) was used for recording HNMR and CNMR spectra at 500 MHz and 125 MHz, respectively, using tetramethylsilane (TMS) as an internal standard. The symbols s (singlet), d (doublet), t (triplet), and m (multiplet) were used for spin multiplicities, and the J values are illustrated in Hertz (Hz). The IR data were obtained using a Nicolet FTIR 550 (Magna) instrument (Nicolet Instrument Corporation, Madison, WI, USA) with KBr disks. Melting points were measured with an Electrothermal IA9200 digital programmable melting point apparatus. The progress of reactions and evaluation of the purity of the compounds were checked with TLC. The partition coefficient values (log P) of the synthesized compounds were predicted using ChemDraw Ultra software version 12.0.
3.1.1. General Procedure for the Synthesis of Compounds 1c, 1d, 2a, 3c, 4b, 5, and 6b
3.1.1.1. 2-(1-Methyl-1H-indazol-3-yl)-4,5-dihydrooxazole (1c)
To a solution of 1-methyl-1H-indazole-3-carbonyl chloride 1a (2 mmol) in acetonitrile (20 mL), bromoethylamine (2.1 mmol) and potassium carbonate (2.5 mmol) were added. The mixture was stirred for 5 hours under reflux conditions and then cooled to room temperature. The crude product was filtered, dried, and crystallized from ethanol to obtain pure compound 1c. Yield: 88%; mp 146 - 148°C. IR (KBr); ν cm-1: 2938, 1660. 1HNMR (500 MHz, DMSO-d6) δ: 8.08 - 8.41 (m, 1H), 7.64 - 7.77 (m, 1H), 7.44 - 7.55 (m, 1H), 7.32 - 7.40 (m, 1H), 3.60 - 4.54 (m, 7H, 2CH2 and NCH3). Mass; m/z (%): 201.2 (18), 181.2 (55), 176.2 (83), 159.2 (78), 151.1 (100), 138.1 (15), 131.1 (34), 116.1 (12), 111.1 (22), 104.1 (23), 89.1 (13), 75.1 (21), 64.1 (9).
3.1.1.2. Ethyl 1-(2-(1-Methyl-1H-indazole-3-carboxamido)ethyl) piperidine-4-carboxylate (1d)
A mixture of 2-(1-methyl-1H-indazol-3-yl)-4,5-dihydrooxazole 1c (2 mmol), ethyl piperidine-4-carboxylate (2.1 mmol), and p-toluenesulfonic acid (2.1 mmol) was refluxed in dry xylene for 48 hours. Water (25 mL) was added and mixed well. The xylene layer was separated, and anhydrous sodium sulfate was added. After filtration, the solvent was completely removed by rotary evaporator, and the resulting oil was purified by flash chromatography (EtOAc: Hexane; 10:90). The purified solid was crystallized in ethanol to obtain pure compound 1d. Yield: 73%; mp 145-147°C. IR (KBr); ν cm-1: 2955, 1705, 1664. 1HNMR (500 MHz, DMSO-d6) δ: 8.30 (d, J = 7.8 Hz, 1H), 8.18 (d, J = 7.8 Hz, 1H), 8.05 (s, 1H, NH), 7.35 - 7.55 (m, 2H), 4.69 (t, J = 7.7 Hz, 2H, OCH2), 4.10 - 4.24 (m, 6H), 4.04 (s, 3H, NCH3), 3.05 - 3.55 (m, 1H), 2.50 - 2.82 (m, 2H), 1.50 - 1.82 (m, 2H), 1.25 (t, J = 7.7 Hz, 3H, CH3). 13CNMR (125 MHz, DMSO-d6): 175.32, 162.81, 162.56, 141.15, 136.89, 134.33, 126.80, 123.65, 122.31, 109.32, 63.96, 63.90, 45.02, 41.17, 38.28, 36.01, 28.59, 27.35, 14.18. Mass; m/z (%): 359.3 (45), 314.3 (55), 170.2 (100), 142.2 (6), 157.2 (8), 142.2 (6), 124.4 (5), 106.4 (6), 77.1 (5).
3.1.1.3. Ethyl 1-(1-Methyl-1H-indazole-3-carbonyl) piperidine-4-carboxylate (2a)
A mixture of 1-methyl-1H-indazole-3-carbonyl chloride 1a (2 mmol), ethyl piperidine-4-carboxylate (2.1 mmol), and potassium carbonate (2.5 mmol) was stirred in dry acetonitrile overnight at room temperature. After evaporation of the solvent, the solid material was separated and dried at room temperature. Crystallization was carried out in ethanol to obtain pure compound 2a. Yield: 83%; mp 165 - 167°C. IR (KBr); ν cm-1: 2955, 1724, 1664. 1HNMR (500 MHz, CDCl3) δ: 8.39 (d, J = 7.5 Hz, 1H), 8.23 (d, J = 7.5 Hz, 1H), 7.25 - 7.55 (m, 2H), 4.15 - 4.32 (m, 2H), 4.04 (s, 3H, NCH3), 4.00 (q, J = 7.5 Hz, 2H, OCH2), 3.11 - 3.65 (m, 1H), 2.50 - 2.82 (m, 2H), 1.50 - 1.82 (m, 2H), 1.27 (t, J = 7.5 Hz, 3H, CH3). 13CNMR (125 MHz, CDCl3): 174.32, 162.80, 162.79, 141.01, 136.81, 126.96, 123.63, 122.23, 109.50, 63.96, 45.02, 41.17, 38.28, 36.01, 28.59, 27.35, 14.18. Mass; m/z (%): 315.1 (35), 270.1 (9), 156.1 (100), 131.1 (16), 82.1 (65).
3.1.1.4. 1-(1-Methyl-1H-indazole-3-carbonyl) piperidine-4-carboxylic acid (2b)
A mixture of compound 2a (2 mmol) was dissolved in 20 mL of ethanol, and 20 mL of sodium hydroxide solution (2N) was added. The mixture was heated at 60°C for 4 hours. The progress of the reaction was monitored by TLC. The pH of the solution was neutralized with 2N hydrochloric acid solution, and the volume of the mixture was reduced to half of the initial level. The pH of the mixture was adjusted to about 3, and extraction was carried out with ethyl acetate. After evaporation of the organic phase, purification was performed by flash chromatography using hexane:ethyl acetate (3:1) as the solvent. Yield: 72%; mp 188 - 191°C. IR (KBr); ν cm-1: 3225, 2955, 1714, 1664, 1580. 1HNMR (500 MHz, CDCl3) δ: 12.36 (bs, 1H, COOH), 7.95 (dd, J = 7.6 and J = 1.5 Hz, 1H), 7.69 (dd, J = 7.6 Hz and J = 1.8 Hz, 1H), 7.40 - 7.50 (m, 1H), 7.20 - 7.29 (m, 1H), 4.46 - 4.55 (m, 2H), 4.14 (s, 3H, NCH3), 3.01 - 3.05 (m, 1H), 1.81 - 1.95 (m, 2H), 1.55 - 1.59 (m, 2H). 13CNMR (125 MHz, CDCl3): 176.04, 162.09, 141.51, 137.91, 126.99, 123.77, 122.08, 121.96, 110.55, 51.69, 45.77, 41.70, 40.73, 28.39, 25.68. Mass; m/z (%): 287.1 (25), 159.1 (65), 142.1 (57), 128.2 (100), 82.1 (17).
3.1.1.5. Ethyl (4-Chlorobenzoyl) glycinate (3b)
Compound 3b was prepared as reported in the literature (CAS No.: 39735-52-9). Yield: 72%; mp 95 - 97°C.
3.1.1.6. N-(2-(Benzylamino)-2-oxoethyl)-4-chlorobenzamide (3c)
A mixture of 3b (2 mmol) and AlCl3 (2 mmol) was stirred in dry acetonitrile (30 mL) for 30 minutes, and then a mixture of triethylamine (2 mmol) and benzylamine (2 mmol) was added and stirred at room temperature overnight. The solid formed was filtered and washed with water. Crystallization was carried out in ethanol to obtain pure compound 3c. Yield: 85%; mp 135 - 137°C. IR (KBr); ν cm-1: 3305, 3276, 2938, 1644, 1596, 1544. 1HNMR (500 MHz, CDCl3) δ: 8.88 (t, J = 7.5 Hz, 1H, NH), 8.45 (t, J = 5.5 Hz, 1H, NH), 7.91 (d, J = 7.5 Hz, 2H), 7.55 (d, J = 7.5 Hz, 2H), 7.17 - 7.35 (m, 5H), 4.30 (d, J = 5.5 Hz, 2H, -NH-CH2-CO-NH-), 3.91 (d, J = 7.5 Hz, 2H, -NH-CH2-Ph). 13CNMR (125 MHz, DMSO-d6): 169.3, 165.95, 139.89, 133.28, 129.79, 128.78, 128.66, 127.61, 127.16, 43.22, 42.80. Mass; m/z (%): 304.1 (4), 302.1 (12), 170.1 (23), 168.1 (69), 139.1 (100), 106.1 (78), 91.1 (45), 75.1 (18), 51.6 (6).
3.1.1.7. 2-(4-Chlorophenyl)-4,5-dihydrooxazole (4a)
A mixture of 4-chlorobenzoyl chloride 3a (2 mmol), bromoethylamine (2.1 mmol), and potassium carbonate (2.5 mmol) was refluxed in dry acetonitrile overnight. After evaporation of the solvent, the solid material was separated and dried at room temperature. Crystallization was carried out in ethanol to obtain pure compound 4a. Yield: 90%; mp 144 - 146°C. 1HNMR (500 MHz, DMSO-d6) δ: 7.85 (d, J = 7.8 Hz, 2H), 7.52 (d, J = 7.8 Hz, 2H), 4.40 (t, J = 7.5 Hz, 2H), 3.95 (t, J = 7.5 Hz, 2H). 13CNMR (125 MHz, DMSO-d6): 162.53, 136.58, 129.93, 129.19, 126.81, 68.05, 54.95. Mass; m/z (%): 181.1 (56), 151.1 (100), 139.1 (15), 125.1 (12), 111 (23), 102 (4), 89.1 (10).
3.1.1.8. Ethyl 1-(2-(4-Chlorobenzamido)ethyl) piperidine-4-carboxylate (4b)
A mixture of 2-(4-chlorophenyl)-4,5-dihydrooxazole 4a (2 mmol), ethyl piperidine-4-carboxylate (2.1 mmol), and p-toluenesulfonic acid (2.1 mmol) was refluxed in dry xylene for 48 hours. Sodium hydroxide solution (2N, 30 mL) was added and mixed well. The organic layer was separated, dried, and anhydrous sodium sulfate was added. The residue was purified by flash chromatography using EtOAc: Hexane (1:4) as the mobile phase. The product was crystallized from ethanol to obtain pure compound 4b. Yield: 62%; mp 156 - 158°C. IR (KBr); ν cm-1: 1724, 1644. 1HNMR (500 MHz, DMSO-d6) δ: 8.46 (t, J = 7.0 Hz, 1H, NH), 7.85 (d, J = 7.5 Hz, 2H), 7.51 (d, J = 7.5 Hz, 2H), 4.06 (q, J = 7.5 Hz, 2H, -OCH2), 3.45 - 3.59 (m, 2H), 3.31 - 3.39 (m, 2H), 2.81 - 2.93 (m, 4H), 2.22 - 2.35 (m, 1H), 1.79 - 1.85 (m, 2H), 1.65 - 1.78 (m, 2H), 1.19 (t, J = 7.5 Hz, 3H, CH3). 13CNMR (125 MHz, DMSO-d6): 174.82, 165.52, 133.71, 129.55, 128.73, 60.25, 57.81, 40.64, 37.38, 28.37, 14.56. Mass; m/z (%): 340.1 (1), 338.2 (3), 295.1 (2), 293.1 (6), 185.1 (2), 183.2 (6), 170.1 (100), 139.1 (12).
3.1.1.9. Benzyl 4-(4-Chlorobenzamido) butanoate (5)
A mixture of 4-chlorobenzoyl chloride 3a (2 mmol), benzyl 4-aminobutanoate (2.1 mmol), and potassium carbonate (2.5 mmol) was stirred in dry acetonitrile overnight at room temperature. The solvent was removed under vacuum, and 20 mL of water and 20 mL of EtOAc were added. The mixture was well mixed, and the organic phase was separated, dried, and filtered. The crude product was purified by column chromatography using EtOAc:hexane (1:5) as the mobile phase. The separated product was an oil. Yield: 62%. IR (KBr); ν cm-1: 1731, 1665. 1HNMR (500 MHz, DMSO-d6) δ: 8.56 (t, J = 7.0 Hz, 1H, NH), 7.87 (d, J = 7.5 Hz, 2H), 7.52 (d, J = 7.5 Hz, 2H), 7.33 - 7.56 (m, 5H), 5.08 (s, 2H, -OCH2), 3.21 - 3.39 (m, 2H), 2.43 (t, J = 7.2 Hz, 2H), 1.79 - 1.85 (m, 2H). 13CNMR (125 MHz, DMSO-d6): 174.29, 165.21, 136.95, 139.89, 133.23, 129.03, 128.33, 128.21, 127.87, 127.81, 127.36, 31.01, 24.38. Mass; m/z (%): 298.1 (10), 196.1 (12), 168.1 (16), 139.1 (25), 122.1 (65), 105.1 (100), 77.1 (12).
3.1.1.10. Ethyl 1-(2-Oxo-2-(4-phenylpiperazin-1-yl)ethyl) piperidine-4-carboxylate (6b)
A mixture of 1-phenylpiperazine 6 (4 mmol), ethyl chloroacetyl chloride (4.1 mmol), and potassium carbonate (4.5 mmol) was stirred in dry acetonitrile (20 mL) at 60°C. The progress of the product was checked by TLC. The crude product (6a) was directly used for the next step. Ethyl piperidine-4-carboxylate (4.1 mmol), potassium carbonate (2.5 mmol), and 30 mL of DMF were added, and the mixture was refluxed in dry DMF for 6 hours. The solvent was evaporated under reduced pressure, and 20 mL of water and 30 mL of EtOAc were added. The organic phase was separated, dried on sodium sulfate, and filtered. The solvent was removed under vacuum, and the crude product was crystallized from ethanol to obtain pure compound 6b. Yield: 82%; mp 156 - 158°C. IR (KBr); ν cm-1: 1724, 1632. 1HNMR (500 MHz, DMSO-d6) δ: 7.22 (d, J = 7.5 Hz, 2H), 6.90 - 6.99 (m, 2H), 6.80 - 6.90 (m, 1H), 4.04 (q, J = 7.5 Hz, 2H, -OCH2), 3.69 (bs, 2H), 3.45 - 3.65 (m, 2H), 2.90 - 3.15 (m, 6H), 2.70 - 2.85 (m, 2H), 2.25 - 2.336 (m, 1H), 2.00 - 2.15 (m, 2H), 1.70 - 1.85 (m, 2H), 1.50 - 1.65 (m, 2H), 1.16 (t, J = 7.5 Hz, 3H, CH3). 13CNMR (125 MHz, DMSO-d6): 174.77, 167.96, 151.32, 129.43, 119.72, 116.27, 61.60, 60.25, 52.53, 49.46, 48.86, 45.48, 41.53, 28.49, 14.55. Mass; m/z (%): 359.1 (6), 314.3 (6.5), 170.2 (100), 142.2 (10), 106.2 (11), 77.1 (12), 44.1 (25).








 agonist derivatives 1d, 2a and 6b")




 on the latency after using pentylenetetrazole (PTZ) in rat in comparisons with diazepam control groups. Animal received test compounds 30 min before PTZ administration. The results are presented as mean ± SEM. The following symbol were used for interpretation of statically aspects: * P < 0.05, ** P < 0.01, *** P < 0.001 compared to vehicle and diazepam.")
")
 conformation: An RMSD assessment")
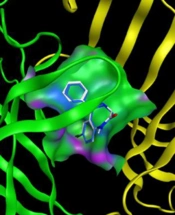
![Superposition of the synthesized compounds on allosteric binding site [diazepam (white) and synthesized compounds (blue)] and corresponding 2D graphical presentation of each ligand interaction with binding pocket’s amino acids](https://brieflands.com/journals/ijpr/articles/157392/figures/ijpr-157392-i009-F9-preview.webp "Superposition of the synthesized compounds on allosteric binding site [diazepam (white) and synthesized compounds (blue)] and corresponding 2D graphical presentation of each ligand interaction with binding pocket’s amino acids")
