

1. Context
Coagulation factor VII (FVII), the first factor in the coagulation cascade, is one of the most critical factors in maintaining hemostasis in physiological conditions. Blood fluidity and vascular system are maintained by primary and secondary hemostasis systems after tissue injury (1). Primary hemostasis is defined as platelet plug formation and platelet aggregation, while secondary hemostasis is insoluble fibrin deposition via the coagulation cascade. During vascular injury, the vessel is immediately contracted at the injury site resulting in platelet plaque formation, fibrin clot generation, white blood cell accumulation in the damaged tissue region, and inflammation and repair initiation (1).
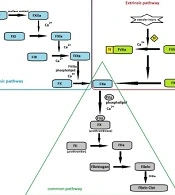
The coagulation cascade is initiated through intrinsic or extrinsic pathways (Figure 1). The extrinsic pathway requires tissue factor (TF), a single transmembrane glycoprotein located in the vascular wall, and becomes exposed to blood only after vascular injury. TF has a high affinity for FVII, and in the presence of calcium ions, these molecules form a complex that initiates coagulation through the extrinsic pathway (2). Although the precise mechanism of blood coagulation is not well defined, the extrinsic pathway seems to play a significant physiological role (3). All the required protein components are available in the blood circulation in the intrinsic pathway, and no external protein source is required. Initiation of intrinsic pathway requires negatively charged surfaces. The intrinsic pathway is clearly important in blood clotting in vitro. However, its physiological significance is unknown, and the activation of this pathway may be limited to non-physiological conditions, such as exposing blood to glass or kaolin (2, 3).
 or vascular injury (through extrinsic pathway). Both pathways lead to activation of FX, where the common pathway begins. Eventually, the conversion of fibrinogen to fibrin culminates in fibrin clot formation, ensuring the maintenance of hemostasis.")
Overview of coagulation cascade: Coagulation cascade initiates by surface contact (through intrinsic pathway) or vascular injury (through extrinsic pathway). Both pathways lead to activation of FX, where the common pathway begins. Eventually, the conversion of fibrinogen to fibrin culminates in fibrin clot formation, ensuring the maintenance of hemostasis.
Coagulation factors include enzymes, non-enzymatic cofactors (such as factors VIII and V, TF, and thrombomodulin), and structural proteins (fibrinogen). All the enzyme proteins involved in coagulation (such as FVII, FIX, FX, and prothrombin) are vitamin K-dependent serine proteases circulating in the plasma in the form of zymogen (inactive precursor), which become activated a few seconds after a vascular injury (2). This review highlights the molecular and physiological properties of coagulation FVII and describes its role in hemostasis. We also discuss the clinical aspects and consequences of FVII deficiency.
2. FVII Protein
Mature FVII (50 kDa-406 amino acids) is a glycoprotein synthesized by the liver cells and secreted into the serum as a serine protease zymogen at a low concentration of 10 nM (500 ng/mL) (4, 5). Spontaneous extracellular proteolytic cleavage of about 1% of the serum FVII in the absence of vascular injury produces activated FVII (FVIIa). Factor seven activating protease (FSAP) is a plasma protein thought to be responsible for activating FVII. However, further studies have shown that the role of FSAP in activating FVII is negligible. Thus, the origin of FVIIa in plasma remains unknown (6-8). The half-life of circulating FVII zymogen in humans is about 4 - 6 hours, while the plasma half-life of FVIIa is 2.5 hours. Compared to other vitamin K-dependent coagulation proteases, the circulating half-life of FVII and FVIIa is very short, complicating the treatment of FVII deficient individuals (Table 1) (9).
| Coagulation Factor | Plasma Half-life |
|---|---|
| Factor FVII | 4 - 6 hours |
| Factor VIIa | 2.5 hours |
| Fibrinogen | 2 - 4 days |
| prothrombin | 3 - 4 days |
| Factor V | 36 hours |
| Factor X | 40 - 60 hours |
| Factor XI | 50 hours |
| Factor XIII | 9 - 12 days |
The Plasma Half-life of Coagulation Factors
FVII has either a 38 or a 60 amino acid signal peptide required for translocating the newly synthesized protein into the lumen of the endoplasmic reticulum. Propeptide, which follows the signal peptide, directs the vitamin K-dependent γ-carboxylation of the mature FVII. This domain is part of the FVII pre-pro leader. Upon transfer to the trans-Golgi, propeptide is cleaved and removed. Propeptide cleavage occurs in the trans-Golgi just before the secretion of FVII. The amino terminus of the mature FVII has a region rich in γ-carboxyglutamic acid (Gla). The precursor FVII has a peptide that directs the γ-carboxylation of 10 glutamic acids. The Gla residues are essential for FVII to obtain calcium-dependent conformation and bind to phospholipid surfaces (10, 11).
The following domains in the FVII structure are the two epidermal growth factor-like (EGFs) domains, followed by an activation domain. EGF domains are responsible for interacting with the cell membranes and other coagulation cascade proteins. The activation domain provides a proteolytic cleavage site. In the presence of calcium ions and phospholipids, the single-chained FVII is quickly hydrolyzed by FXa (activated FX) and thrombin and converted into a two-chained form (6, 11). Activation of FVII involves cleavage of the peptide bond between Arg152 and Ile153 in the activation domain creating a heavy and a light chain that remain attached by disulfide bonds. This proteolysis is associated with an 85-fold increase in FVII activity. The remainder of the protein comprises the serine protease catalytic domain. Vitamin K-dependent coagulation factors, including FVII, have conserved disulfide bonds. There are three disulfide bonds in each EGF domain and several disulfide bonds in the catalytic domain of FVII. Disulfide bonds form in the endoplasmic reticulum, and chaperons such as protein disulfide isomerase (PDI) are essential for forming proper disulfide bonds (6, 10, 12).
FVIIa has catalytic activity but acts more as a zymogen before binding to TF. The FVIIa-TF complex formation increases the catalytic activity of FVIIa, by more than 1000 fold (13). It has been suggested that TF probably causes a change in the active site of FVIIa, which leads to a more efficient hydrolysis of the substrate. The interaction of FVIIa with TF depends on calcium ions. The Gla domain saturation with Ca2+ is likely responsible for the high affinity of FVIIa for TF (14). Various studies have shown non-hemostatic properties for TF and FVIIa-TF complex, such as cell signaling, metastasis, and angiogenesis (15-18).
3. Factor VII Gene
The human FVII encoding gene (F7) is a single copy gene on the long arm of chromosome 13 (13q34) and is 2.8 kb downstream of the FX (F10) gene (6). O'Hara et al. reported the complete nucleotide sequence of the human F7 gene in 1987 (19). The F7 gene (14.8 kb) includes nine exons and eight introns (Table 2). Exon 1a, 1b, and part of exon 2 code for a pre-pro leader sequence that is cleaved away during the protein processing. The remaining exons code for 406 amino acids that remain in the mature protein circulating in the blood (19). The F7 gene gives rise to three different mRNA transcripts. The first transcript (NM_019616.4) codes for a protein with a 38 amino acid long pre-pro leader (total length of the protein is 444 residues), and the second transcript (NM_000131.4) codes for an FVII molecule with a 60 amino acid long pre-pro leader (total length of the protein is 466 residues). Genomic sequence information has shown that the first transcript lacks an optional exon (exon 1b). Both transcripts are functional and give rise to the production of biologically active FVII. The final product of both transcripts (mature FVII protein) is identical. In the normal liver, mRNA lacking exon 1b is more abundant than the mRNA possessing this exon. The third transcript (NM_001267554.1) lacks the amino-terminal domains. However, the physiological significance of the third transcript is unknown (6). As for other vitamin K-dependent proteins, each of the exons of the F7 gene codes for a separate protein domain. These domains are conserved between vitamin K-proteins, and between them, the activation domain has the lowest degree of conservation (19).
| Exon | Intron | Coding Region | Length (bp) |
|---|---|---|---|
| 1a | Pre-pro leader | 100 | |
| 1a | 1068 | ||
| 1b | Pre-pro leader | 66 | |
| 1b | 2574 | ||
| 2 | Pre-pro leader, Gla domain | 161 | |
| 2 | 1928 | ||
| 3 | Gla domain | 25 | |
| 3 | 70 | ||
| 4 | EGF domain | 139 | |
| 4 | 1716 | ||
| 5 | EGF domain | 141 | |
| 5 | 971 | ||
| 6 | Activation domain | 110 | |
| 6 | 595 | ||
| 7 | Catalytic domain | 124 | |
| 7 | 817 | ||
| 8 | Catalytic domain | 1622 |
The Exons and Introns of the F7 Gene
Promoter and other regulatory elements located immediately 5' to the human F7 gene have been extensively investigated. The promoter region of the F7 gene lacks the typical TATA-box. This feature is also seen in the promoter of other coagulation proteins such as FXII, FX, FIX, and prothrombin. Although a CAAT-box is critical for FIX and FX promoters, there is no such element in the FVII promoter region. The major transcription initiation region of the F7 gene is located within a strong promoter element at position -51 upstream of the translation starting point. Sp1 and HNF-4 binding sites, which are crucial in the promoter activity of F7, are located upstream of the transcription start site (Figure 2). There are two silencer elements upstream of the promoter region (20). In addition, the F7 gene can be regulated at least by a remote gene. Fagan et al. have shown that at least a gene locus on 8p23.2-23.1 controls FVII levels (21).

Major transcription binding sites and promoter elements of the F7 gene
4. F7 Gene Variations
A significant number of mutations have been reported in the F7 gene. According to the FVII gene variant database (factorvii.org) and human gene mutation database (hgmd.cf.ac.uk), there are more than 200 different variants throughout the F7 gene that can affect all protein domains. Missense mutations are the most frequent type of mutation, followed by splice site mutations, promotor mutations, nonsense mutations, and small insertions and deletions (15). Many of these mutations are considered the cause of FVII deficiency, but only a small number of them have been functionally studied and described. The International Registry on FVII deficiency (IRF7) and the Greifswald Registry record more than 1000 FVII deficiency genetic diagnoses (22, 23). In the F7 gene, several large rearrangements have been identified that can lead to FVII deficiency. So far, five large chromosomal deletions of chromosome 13, including the entire or part of the F7 gene, have been reported (24-27). Despite the observation of mutations in the promoter, exon, and splice sites in most patients, less than 10% of the patients had no mutations in the F7 gene. Even with next-generation sequencing, mutations in some patients remain unknown, suggesting that FVII deficiency could be caused by mutations in genes other than F7 (28, 29).
The plasma levels of FVII (activity and antigen) are variable between healthy and FVII deficient subjects, making FVII:C and FVII:Ag tests (FVII antigen level) ineffective for diagnosing FVII deficiency. Different environmental factors such as sex, age, weight, and diabetes are of considerable importance. For example, FVII levels increase with age and are lower in women than men at younger ages (30). On the other hand, differences in plasma levels of FVII can also be due to genetic factors in which the role of multiple polymorphisms has been proven (31).
G > A substitution at position -402 and G > T in -401 are two common unrelated functional polymorphisms in the promoter region of the F7 gene. The -401T allele decreases the basal transcription level of the F7 gene. In contrast, the -402A allele is associated with increased transcriptional activity of the F7 gene (32). The FVII levels in healthy subjects are reduced by 25% when a 10bp insertion (rs36208070) occurs at position -323 in the 5'UTR of the F7 gene (20). The G73A polymorphism (rs6039) in intron 1a of the F7 gene can also affect the FVII level. The A73 allele reduces the plasma levels of FVII (33). Previous studies revealed that coding region polymorphisms could also affect the FVII plasma level. An example is the Q353 allele of R353Q (rs6046) polymorphism derived from the substitution of G to A at position 10976 of exon 8, which causes a 25% decrease in FVII activity and antigen levels (34). Additionally, genome-wide association studies (GWAS) have identified several genomic regions associated with FVII levels that may provide additional data on how FVII levels vary among individuals (35, 36).
Multiple studies have investigated the association between F7 polymorphisms and cardiovascular diseases. In this regard, a meta-analysis revealed that the -323Ins10 polymorphism was significantly associated with coronary heart disease (CHD) in Asians and Europeans, and R353Q polymorphism showed an association with CHD in the Asian population (37). Another meta-analysis confirmed the association of R353Q with CHD and suggested that R353Q polymorphism was associated with the reduced risk of CHD in Asians (38). F7 gene polymorphisms may be involved in response to anticoagulants such as warfarin. Research has shown that R353Q plays a critical role in the initial response to warfarin (39).
For some mutations detected in patients, recombinant FVII expression and site-directed mutagenesis of F7 have been used in various functional studies. These studies examine the effects of different variants on FVII features such as the secretion rate, ligand binding, coagulation activity, and intracellular localization to elucidate the molecular basics of the deficiency. FVII mutants are valuable tools for evaluating single residues or defining essential regions involved in the structure-function relationship and the formation of macromolecular complexes (40). Site-directed mutagenesis, in vitro expression, and description of TF and FVII variants have identified essential amino acids for TF binding and TF-FVIIa activity. Such studies suggest that the binding of FVII to TF occurs through a large interface, and the interface includes four FVIIa domains and two extracellular TF domains (41). A study showed that FVII with the R79Q mutation was normally expressed but had a reduced affinity for TF binding (42). In this regard, further studies taking advantage of X-ray crystallography showed that the side chain of this residue played an important role in the interaction of EGF1 with TF (43). Elsewhere, an investigation indicated that the F328S mutation reduced the FVII affinity for TF and prevented FVII from activating FX, possibly due to a disruption in the site of attachment to the substrate (44). The R152Q mutation showed poor expression in another functional study and had no detectable coagulation activity. This mutation occurs at the site of proteolytic cleavage of FVII and inhibits the activation of FVII (45). H348R and S282R mutations detected in a compound heterozygote patient were examined in a study. Both mutations showed reduced secretion and coagulant activity while not altering the protein's intracellular localization (40).
Some mutations lead to intracellular accumulation of FVII. For instance, the T359M mutation causes FVII to accumulate within the cell and thus results in a severe defect in F7 secretion (46). Interestingly, another study examining the functional properties of FVII showed that C91S mutation led to increased protein secretion in the culture medium while severely reducing coagulant activity (5).
Cysteine residues play an essential role in the function and structure of FVII protein. For instance, the Cys329Gly mutation in the catalytic domain disrupts the formation of a disulfide bond with Cys310. This disulfide bond is essential for binding TF and the catalytic function of FVIIa (47).
The development of advanced bioinformatics software and in silico tools enables us to investigate the phenotype-genotype correlation and predict the effects of novel mutations detected in genetic diseases, including FVII deficiency. Tiscia et al. characterized a novel variation (c.1199G>C) using the bioinformatics tools such as PROMO, SIFT, and Polyphen-2. In silico predictions revealed that c.1199G>C had a damaging effect on FVII conformation via influencing the formation of the Cys400-Cys428 disulfide bond (48).
Novel variations keep being detected, and their effects need to be examined using in silico and laboratory testing. In 2021, Zhang et al. detected four novel variations (c.251T>C, c.466G>A, c.1016C>T, c.‐16T>G) and investigated their pathogenicity using PyMOL2.4, Swiss‐PDB Viewer, SIFT, POLYPHEN‐2. Bioinformatics analyses found these variations pathogenic (49). In another study, Liang et al. detected two novel variations in three patients with FVII deficiency; the molecular model analysis of the two novel mutations (Cys115Arg and Pro324Leu) indicated impairment of the proper folding of the EGF1 domain and impairment of the F7 coagulant activity (50). Cys164Tyr, another novel mutation, was found in a patient with mild clinical manifestations despite deficient FVII activity (51).
5. FVII Deficiency
Coagulation defects other than Hemophilia A, Hemophilia B, and von Willebrand's disease, generally autosomal recessive, are rare, and the frequency of homozygous individuals in the general population ranges from 1 in 2,000,000 for FII and FXIII deficiencies to 1 in 500,000 for FVII deficiency (9, 52, 53). There are some exceptions where these deficiencies have higher frequencies, such as countries with large Jewish populations, the Middle East countries, and South India. In the latter two cases, familial marriages are relatively common and cause autosomal recessive traits to become more abundant (54, 55).
Congenital FVII deficiency (OMIM 227500) is the most common rare congenital coagulation disorder (RICD) (56); it has autosomal recessive inheritance and is often associated with consanguineous marriage. Heterozygous individuals have approximately 50% of normal coagulation factor levels and usually lack clinical symptoms, but this is not always the case, and occasionally, a heterozygote may have significant bleeding events. In such cases, the coexistence of a polymorphism affecting the FVII level might be the underlying cause (31, 57). Based on activity and antigen levels, FVII deficiency is divided into three categories: CRM- (cross-reacting material negative), CRM+, and CRMred (58).
Clinical manifestations of congenital FVII deficiency vary widely, ranging from asymptomatic to life-threatening bleeding (59). Based on clinical features, people suffering from FVII deficiency are categorized into severe, mild/moderate, and asymptomatic groups. There is significant molecular and phenotypic heterogeneity in FVII-deficient individuals. Box 1 summarizes the clinical manifestations of FVII deficiency. The frequency of symptomatic females is more than symptomatic males due to menorrhagia, which is the most common symptom in women. Life-threatening bleeding, including gastrointestinal bleeding and central nervous system hemorrhage (in 20% of patients), is seen in people with a lower activity level of FVII. Life-threatening bleedings often (70% of cases) occur in the first six months of life and are associated with high mortality rates. The most significant risk factor for CNS bleeding is the trauma that occurs during childbirth (6, 60, 61).
| Asymptomatic |
| Mild (having one or two of the following symptoms) |
| Moderate (having three or more of the following symptoms) |
| Mucous membrane bleeding, including epistaxis and oral bleeds |
| Non-life threatening gastrointestinal bleeding |
| Heavy menstrual bleeding |
| Bleeding during or after surgery |
| Joint or significant soft tissue bleeds and cutaneous bleeds following identifiable trauma |
| Sever |
| Mucous membrane bleeding that is life-threatening or that requires RBC transfusion |
| Life-threatening gastrointestinal bleeding, particularly intra-mural bleeds or where no lesion is identified |
| Recurrent Joint or significant soft tissue bleeds |
| CNS bleeds |
| Spontaneous ocular bleeding |
The Clinical Manifestations of Congenital FVII Deficiency
5.1. Severe Factor VII Deficiency
People with severe FVII deficiency may die due to bleeding at or shortly after birth. Severe cases are homozygous or compound heterozygotes for deleterious mutations, resulting in FVII:C of less than 2%. Most mutations disrupt the expression, including promoter mutations, splice-site mutations, or frameshift mutations (61).
5.2. Mild/Moderate Factor VII Deficiency
Cases with mild or moderate clinical phenotypes are homozygous or compound heterozygotes for deleterious mutations. FVII:C measured in these individuals varies from less than 1 to 52% and does not correlate well with the clinical severity of symptoms. Differentiating between mild and severe cases based on FVII:C or FVII antigen (FVII:Ag) measurement is impossible (61). Coinheritance of FV Leiden with FVII deficiency enhances thrombin formation and leads to mild bleeding phenotypes (6).
5.3. Asymptomatic Cases
Asymptomatic subjects have FVII:C from 4 to 61%, and FVII:Ag from 5 to 113%. All known mutations in these individuals are missense substitutions. The diagnosis of asymptomatic cases is usually made during family studies or hemostatic screenings (6, 61).
The severity of the clinical phenotype of congenital FVII deficiency reflects the pivotal role of FVII at the onset of coagulation so that a complete lack of FVII in humans results in perinatal mortality (6). Occasionally, deficiencies in the coding genes of proteins that contribute to the post-translational alterations of coagulation factors and vitamin K metabolism may lead to a combined coagulation factors defect (62). One of these cases is the combined defect of FVII and prothrombin (63). Chromosome 13 deletions can also result in the coexistence of FVII and FX deficiencies (64).
In addition to heredity, FVII deficiency is occasionally acquired by the presence of a tumor, liver failure, vitamin K deficiency, or treatment with vitamin K antagonists. Acquired FVII deficiency may also occur due to bone marrow transplantation and bacteremia. Clinical manifestations of acquired FVII deficiency are usually mild to moderate, which may become relevant in the case of surgery (56, 65-67).
5.4. Diagnosis
In the case of congenital and acquired FVII deficiencies, the diagnosis is based on the isolated prolonged prothrombin time (PT) and normal activated partial thromboplastin time (aPTT) (55, 68). FVII:C measurement is necessary to confirm the diagnosis. ELISA technique using monoclonal FVII-specific antibodies can also be used to measure the plasma level of the FVII antigen (68). In patients with severe, mild/moderate, and asymptomatic congenital FVII deficiency, the levels of FVII:Ag and FVII:C are widely overlapping and cannot be used to predict the clinical severity of the disease; thus, collecting a detailed family history is essential (51). However, the FVII:Ag and FVII:C assays allow one to distinguish between CRM- (FVII:Ag and FVII:C reduced with the same ratio), CRM+ (a decreased FVII:C with normal FVII:Ag), and CRMred (FVII:Ag reduction but not as much as FVII:C) (5, 6, 69). The lack of correlation between clinical phenotypes and the laboratory findings probably reflects that only trace amounts of FVII are sufficient to initiate the coagulation process, and routine laboratory assays cannot detect these amounts. Both experimental and mathematical modeling studies have shown that 5 pmol/L of FVIIa may be sufficient for coagulation induction (58).
Genotyping is another way to diagnose RICDs, including FVII deficiency, whose main application is a prenatal diagnosis but should be restricted to families with a history of severe bleeding episodes. Total gene sequencing is the recommended method for genotyping. This method enables us to detect 90 to 92% of mutated alleles (54, 68).
5.5. Treatment
As with hemophilia, replacing a deficient coagulation factor is the main treatment for FVII deficiency (70). Prophylaxis is also essential for patients with severe clinical manifestations and must be considered from childhood or immediately after the first bleeding episode. However, due to the short half-life of FVII, prophylaxis in FVII deficiency demands much effort (71). Pregnancy complicates the management of FVII deficient individuals. During pregnancy, the FVII plasma level increases in the normal population and heterozygote women with the highest threshold in the third trimester. Thus, the highest risk of bleeding may be during the early stages of pregnancy. No FVII level increase has been reported in homozygotes. Therefore, the patient's bleeding history, third trimester PT, FVII level, mode of delivery, and multiple gestations are necessary to manage FVII deficient pregnant women (72, 73). Acquired FVII deficiency is often associated with an underlying pathology, and treating the causative pathology leads to the normalization of FVII activity level. Therapeutic options for congenital and acquired FVII deficiency are the same (68). Available therapeutic options for FVII deficiency are as follows:
5.5.1. Prothrombin Complex Concentrate (PCCs)
The main advantage of this product is the lower volume of injection, fewer allergic reactions, and the possibility of taking strategies to inactivate viruses during production. Because the half-life of FVII is much shorter than other coagulation factors, injection of multiple PCC doses may increase other factors and the risk of thrombotic events (74).
5.5.2. Plasma-Derived FVII Concentrate (pdFVII)
Concentrates of FVII are obtained from pools of plasma. These products are used for preventive treatment and control of serious bleeding and bleeding during surgery. However, plasma-derived concentrates are associated with the risk of transmitting pathogens (74).
5.5.3. Fresh Frozen Plasma
Fresh frozen plasma(FFP) containing all coagulation factors is relatively inexpensive and widely available, but its effectiveness is limited because of the high risk of fluid overload due to repeated infusions and the need for slow infusions. The transmission of viral infections such as hepatitis or HIV viruses is one of the risks of using FFP (6, 74).
5.5.4. Activated Recombinant Factor VII
Activated recombinant factor VII (rFVIIa) is used to treat bleeding events in patients with FVII deficiency and prevent bleeding in patients under surgery (75). Prophylactic administration of rFVIIa has been previously scheduled for children with severe congenital FVII deficiency (76). This product does not contain human plasma and albumin; therefore, there is no risk of human viral transmission. The ideal replacement therapy for patients with FVII deficiency is rFVIIa, but this treatment is costly and not available to all patients (54, 69). Alloimmunization against exogenous FVII, a scarce phenomenon, is the main challenge of this kind of replacement therapy and results in reducing rFVIIa effectiveness (53). Perioperative hemorrhage in neonatal cardiac surgery is a significant cause of morbidity and mortality. Since neonates have immature coagulation cascade and low levels of coagulant proteins, administration of rFVIIa may be a prophylactic option during surgery (77). Researchers are currently conducting various strategies to improve rFVII half-life or activity. So far, some rFVII molecule with improved features has been produced, and clinical trials are in progress (78-80).
6. Conclusions
The FVII protein is a key component of homeostasis, and along with TF, it plays a role in other cellular processes besides blood coagulation. Due to its importance, extensive studies have been performed on the F7 gene, FVII protein, and related phenotypes to determine its molecular and clinical features. So far, researchers have achieved significant results leading to advancements in treating FVII deficiency and improving the quality of patients' lives. These studies have also paved the way to prevent and prenatally diagnose FVII deficiency. Plenty of functional studies have been and are being performed on previously and newly identified mutations to elucidate the phenotype-genotype correlation of FVII deficiency. Despite extensive studies, the genotype-phenotype correlation in patients with FVII deficiency has not yet been fully understood, and further studies are needed to elucidate all aspects of the disease entirely.
