



1. Background
Influenza A is a viral respiratory illness that can lead to pneumonia and severe complications, including acute respiratory distress syndrome. It is responsible for an annual global mortality rate of approximately 290,000 to 650,000 individuals and is known to induce alterations in the secondary structure of proteins (1). The genome of Influenza A consists of a single-stranded RNA molecule with a negative-sense orientation, segmented into eight distinct parts. These segments encode proteins essential for viral replication and pathogenicity (2, 3).
The first three segments and the fifth segment encode components of the viral replication machinery, including nucleoprotein (NP), polymerase A protein (PA), polymerase B1 protein (PB1), and polymerase B2 protein (PB2). Additionally, some strains encode the PB1-F2 protein within the PB1 segment. The fourth and sixth segments encode neuraminidase and hemagglutinin, the surface glycoproteins of the virus. The seventh segment encodes the M1 and M2 proteins, while the smallest segment, NS, encodes the NS1 protein (an interferon antagonist) and NS2 protein from spliced and unspliced mRNA transcripts (4).
In recent decades, several antiviral agents have been developed to treat influenza, including RNA polymerase inhibitors (e.g., Fluorodeoxyuridine analogues, Favipiravir), neuraminidase inhibitors (e.g., Zanamivir, Oseltamivir, Laninamivir, Peramivir), and M2 ion channel inhibitors (e.g., Amantadine, Rimantadine) (5). However, the overuse of M2 ion channel inhibitors has led to widespread resistance in many influenza strains, complicating their clinical application (6-8). Neuraminidase inhibitors have been effective in alleviating clinical symptoms and reducing disease duration for both Influenza A and B viruses. Nevertheless, resistance to oseltamivir is increasingly reported (8) Polymerase inhibitors, such as Fluorodeoxyuridine analogues and Favipiravir, have shown potential in vitro and in vivo, though more clinical research is needed (9)
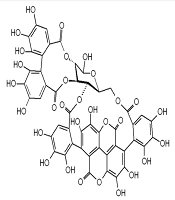
The resistance and adverse effects associated with chemical antiviral drugs have prompted a growing interest in exploring new antiviral agents, particularly herbal-based therapies (6) Punicalagin, a bioactive ellagitannin compound found in pomegranate (Punica granatum), has demonstrated antibacterial (10, 11), antiviral (5, 12-18), and anti-inflammatory properties (11, 13, 19-22) across various studies. Despite evidence supporting its anti-influenza effects, the precise mechanism by which punicalagin inhibits influenza virus replication remains unclear. Simulation studies provide comprehensive insights into drug-protein interactions, offering a pathway to further elucidate these mechanisms (Figure 1).

The chemical structure of Punicalagin
2. Objectives
Given the promising anti-influenza effects of punicalagin demonstrated in previous research, this study aims to deepen our understanding of its mechanism of action by evaluating its inhibitory effects on all influenza virus proteins within a simulated environment.
3. Methods
3.1. Preparation of Molecular Structures
The molecular information and two-dimensional structure of Punicalagin (PubChem CID: 348276747) were obtained from the NCBI PubChem compound database. The three-dimensional structure of Punicalagin was subsequently generated and converted into a PDB file using Avogadro software, followed by optimization and energy minimization. FASTA sequences for the proteins were retrieved from the UniProt.org server, and their PDB files were created using the I-TASSER server. Molecular docking and simulation studies were performed after the optimization and energy minimization processes.
3.2. Protein Molecular Dynamics Simulation
The molecular dynamics simulation was conducted on the PB1-F2 protein alone in water and salt to reflect changes in temperature, pressure, and a 140 mM ion concentration, achieving equilibrium. The PB1-F2 protein was modeled using Gromacs 2021 software with the G43A1 force field, and the simulation was run for 100 nanoseconds using the SPC216 water model (23). Na+ and Cl− ions were added to achieve the target concentration of 140 mM. Energy minimization and simulation were carried out using the steepest descent method, integrating over 50,000 steps. The LINCS algorithm was employed for equilibration under NVT and NPT ensembles. Structural features of the neuraminidase protein in the presence of the ligand were analyzed using the saved simulation trajectories (24). The resulting PDB file was subsequently used for molecular docking studies.
3.3. Molecular Docking
Molecular docking was conducted using AutoDock v.4.2 software on a Linux operating system. A grid box with dimensions of 1.4 × 1.8 × 1.9 nm (x × y × z) was defined for all blind docking experiments. PDBQT files were generated for Punicalagin (as the ligand) and the influenza A virus proteins. Docking was performed over 200 independent runs, and the resulting data were analyzed. Discovery Studio software was utilized to determine the frequency of interactions between the amino acid residues of the virus proteins and Punicalagin, as well as to calculate the types and numbers of amino acids involved in the binding site (25).
3.4. Molecular Dynamics Simulation of Protein-Ligand Complex
Finally, molecular dynamics simulation of the PB1-F2 protein-Punicalagin complex was conducted in an aqueous environment, alongside a simulation of the PB1-F2 protein alone without the ligand (24, 26). The simulation of the PB1-F2 protein-Punicalagin complex was performed for the protein with the lowest binding energy in the presence of the Punicalagin inhibitor. The results were compared to assess structural differences in the secondary structure of the PB1-F2 protein when bound to the ligand versus when unbound. Data were extracted using Excel software and statistically analyzed using paired t-tests and SPSS version 22.
4. Results
4.1. Molecular Docking
Molecular docking results revealed that punicalagin exhibited a higher binding affinity for the PB1-F2 and M2 proteins (Table 1). The estimated free binding energy (BE) of punicalagin with the PB1-F2 protein was -5.76 kcal/mol, and the estimated inhibition constant (Ki) was approximately 60.13 μM (Table 1 and Figure 2). This low binding energy, coupled with a significant number of interactions and a relatively low drug concentration, indicates a strong affinity of punicalagin for the PB1-F2 protein's binding site.
| Variables and Proteins | UniProt ID | BE (kcal/mol) | EIC (µM) | Interaction Bonds |
|---|---|---|---|---|
| HA | P03452 | -2.03 | 32750 | Thr42, Ser339, Ser29, Val41, Ile337, Pro338, Asn28, Asn40, Thr30 |
| M1 | P03485 | -3.78 | 1700 | Ser161, Gin158, Ile51, Il14, Ser157, Pro16, His162, Ile154, Ser53, Thr56, Ser17 |
| M2 | P06821 | -5.10 | 182 | Gly61, Ser64, Glu66, Ser71, Gly62, Leu59, Ala27, Ile28, Gly67, Pro63, Met72, Phe55, Asn31, Asp24 |
| NA | P03468 | -4.31 | 688 | Arg158, Ser157, Arg115, Thr65, Lys63, Asn194, Asn156, Trp61, Leu112 |
| NP | P03466 | -3.71 | 1920 | Arg400, Gln399, Asp439, Ile406, Ser402, Arg446, Ala428, Glu443, Ala401, Ala403, Gln405, Gly404, Thr442 |
| NS1 | P03496 | -4.17 | 874.27 | Glu142, His169, Leu166, Ser165, Ser87, Thr143, Gly168, Pro167, Leu163, Pro164, Pro162, Ile145, Met98 |
| NS2 | P03508 | -3.85 | 1510 | Glu63, Leu55, Arg66, His56, Met50, Glu67, Gly70, Arg51, Glu74, Gln71, Leu58, Gln59 |
| PA | P03433 | -3.30 | 3780 | Trp559, Leu606, Leu580, Lys583, Phe650, Gln610, Gln531, Trp646, Arg613, Val609 |
| PB1 | P03431 | -4.22 | 813.35 | Val644, Gln127, Ala140, Gly125, Ile517, Pro138, Ser515, Pro668, Met646, Ala652, Ala139, Ala643 |
| PB1-F2 | P0C0U1 | -5.76 | 60.13 | Gln19, Gln25, Lys29, Leu77, Trp80, Val76, Lys73, Ile16, Leu30, Lys20, His15, Ser12, Thr74 |
| PB2 | P03428 | -2.95 | 6900 | Ser634, Arg630, Ser635, Phe633, Ile710, Gln632, Asn711, Met631, Tyr592, Ile588 |
Molecular Interaction of Punicalagin with Different Influenza Proteins

The structures and binding sites of Punicalagin in PB1-F2 influenza A protein and amino acid residues of proteins interacted with Punicalagin in the binding site.
For the M2 protein, punicalagin's binding energy (BE) was -5.10 kcal/mol, and the estimated inhibition constant (Ki) was around 182 μM (Table 1). Most interactions were localized in the C-terminal regions of the protein, with only two interactions occurring in the transmembrane domain (Table 1). These findings suggest that punicalagin has a higher affinity for the PB1-F2 protein compared to the M2 protein, as evidenced by the more favorable binding energy and lower inhibition constant associated with PB1-F2.
4.2. Molecular Dynamics
Root mean square deviation (RMSD) analysis indicated that the PB1-F2 protein reached equilibrium in its unbound state at approximately 10 nanoseconds. In contrast, the PB1-F2 protein bound to punicalagin achieved equilibrium at approximately 30 nanoseconds. The simulation system remained stable throughout the 100-nanosecond duration (Figure 3A). Radius of gyration (Rg) calculations revealed that equilibrium was maintained for the PB1-F2 protein in both its unbound state and when bound to punicalagin during the simulation. However, the Rg value decreased when the protein was bound to punicalagin, suggesting increased protein compactness and structural alterations due to binding. This compression may potentially affect the protein's function (Figure 3B).
; B, radius of gyration (Rg); C, changes in hydrogen bonds (H-bonds); D, root mean square fluctuation (RMSF). Black represents the PB1-F2 enzyme in the unbound state, while red denotes the PB1-F2 enzyme bound to punicalagin.")
The amount of molecular dynamics parameters during the 100 nanoseconds of the simulation time related to the binding of punicalagin to the PB1-F2 protein. A, root Mean Square Deviations (RMSD); B, radius of gyration (Rg); C, changes in hydrogen bonds (H-bonds); D, root mean square fluctuation (RMSF). Black represents the PB1-F2 enzyme in the unbound state, while red denotes the PB1-F2 enzyme bound to punicalagin.
Furthermore, the binding of punicalagin to the PB1-F2 protein led to a reduction in the number of intramolecular hydrogen bonds, which likely decreased the protein's dynamic behavior. This reduction could signify an inhibition of the protein's function (Figure 3C). Root mean square fluctuation (RMSF) analysis for individual amino acid residues showed that the binding of punicalagin resulted in reduced fluctuations in residues 15 - 34 and 70 - 80 of the PB1-F2 protein (Figure 3D). The molecular dynamics simulation data for the PB1-F2 protein are summarized in Table 2.
| Variables and Proteins | TE (kJ/mol) | RG (nm) | RMSD (nm) | RMSF (nm) | H-bond |
|---|---|---|---|---|---|
| PB1-F2 | -253790 ± 1320 | 1.3 ± 0.042 | 0.76 ± 0.059 | 0.286 ± 0.13 | 66.6 ± 4.5 |
| PB1-F2-punicalagin | -258055 ± 1126 | 1.25 ± 0.049 | 0.74 ± 0.11 | 0.246 ± 0.15 | 62.46 ± 4.3 |
| P-value c | < 0.001 | < 0.001 | < 0.001 | < 0.001 | < 0.001 |
Table 3 provides a detailed comparison of the secondary structural changes in the PB1-F2 protein during the 100-nanosecond simulation period, both in its unbound state and when complexed with punicalagin. The data indicate a notable reduction in the proportion of coil structures and a corresponding increase in turn structures upon binding with punicalagin. Since secondary structure plays a crucial role in maintaining protein stability and function, these alterations are likely to significantly impact the protein's activity. The observed structural changes suggest that punicalagin binding may hinder the functional efficacy of the PB1-F2 protein by inducing conformational modifications critical to its role.
| Secondary Structure | PB1-F2 | PB1-F2-Punicalagin | ||
|---|---|---|---|---|
| Mean ± SD | % | Mean ± SD | % | |
| Coil | 15.2 ± 3.6 | 17.5 | 13.11 ± 1.4 | 15.1 |
| Bend | 8.36 ± 2.1 | 9.6 | 8.2 ± 2.37 | 9.4 |
| Turn | 11.9 ± 3.69 | 13.6 | 14.05 ± 3.5 | 16.2 |
| α | 51.5 ± 10.5 | 59.2 | 51.45 ± 8.1 | 59.27 |
Secondary Structure Changes Within 100 Nanoseconds of the Simulation Time for the PB1-F2 Molecule in the State Without Binding to Punicalagin and Binding to Punicalagin
5. Discussion
This study aimed to elucidate the inhibitory mechanisms of punicalagin on influenza virus proteins through molecular dynamics and docking analyses. Our findings indicate that punicalagin exhibited significant binding affinity for the PB1-F2 and M2 proteins, with the strongest inhibitory potential observed for the PB1-F2 protein. The high binding energy (BE) and low Estimated Inhibition Constant (EIC) values suggest that punicalagin binds more effectively to PB1-F2 compared to other influenza virus proteins.
PB1-F2, a short viral protein encoded by the PB1 gene, consists of approximately 90 amino acids and is divided into C-terminal (amino acids 65 - 87) and N-terminal (amino acids 1 - 37) domains (27) Previous research has highlighted PB1-F2's role in enhancing the host's inflammatory response during influenza infection through activation of the NF-κB pathway, granulocyte chemotaxis, and apoptosis of immune cells (28) The C-terminal domain is known for its pro-inflammatory activity,(29) while the N-terminal domain contributes to increased PB1 protein expression (30) The results of our study suggest that punicalagin binds to the C-terminal region of PB1-F2, potentially inhibiting pro-inflammatory cytokine production and modulating the inflammatory response.
Punicalagin also demonstrated binding to the M2 protein, with a BE of -5.10 kcal/mol and an EIC of 182 μM. The M2 protein, a proton channel composed of four monomers, plays a crucial role in maintaining pH balance across viral membranes during entry and maturation. It is essential for viral replication and is targeted by influenza drugs such as amantadine and rimantadine (31). The interaction of punicalagin with the M2 protein involves key residues, such as Gly61, Ser64, Glu66, and Ser71 through hydrogen bonds, and Gly62, Leu59, Ala27, Ile28, Gly67, and Pro63 through hydrophobic interactions. Despite these interactions, the removal of the C-terminal helix of M2 does not significantly impact proton conduction, suggesting that punicalagin’s binding may not substantially affect viral replication. However, further in vitro studies are required to confirm these findings (32, 33).
Previous studies have validated the antiviral and anti-influenza effects of pomegranate extracts and punicalagin. Sundararajan et al. reported that pomegranate polyphenols inhibit hemagglutinin and exert direct or indirect effects on the viral envelope, preventing its fusion with host cell membranes (34). Salles et al. found that pomegranate ethanol extracts and punicalagin exhibit virucidal effects against the Mayaro virus by damaging viral particles (35). Haidari et al. demonstrated that pomegranate polyphenols inhibit hemagglutinin activity and exert direct antiviral effects (17) while Li et al. highlighted punicalagin's role in inhibiting neuraminidase (NA) activity (18).
In our study, punicalagin bound to the NA protein with a BE of -4.31 kcal/mol and an EIC of 688 μM, forming six hydrogen bonds and two hydrophobic interactions. This interaction aligns with previous findings of punicalagin’s inhibitory effect on NA activity. Molecular dynamics simulations revealed significant structural changes in the PB1-F2 protein upon punicalagin binding, including alterations in its radius of gyration, secondary structure, and intramolecular hydrogen bonds. These structural modifications likely impact the protein’s functionality, further supporting the potential of punicalagin as an effective antiviral agent.
Overall, the study underscores the therapeutic potential of punicalagin against influenza by targeting key viral proteins and modifying their functional dynamics. However, this research is based on in silico models, and the findings need validation through experimental studies. The predictive nature of molecular docking and dynamics simulations suggests that while punicalagin exhibits strong binding affinities, these predictions must be corroborated through biological investigations, including in vitro and in vivo experiments.
5.1. Conclusions
Punicalagin demonstrated the highest binding affinity for the PB1-F2 protein among the influenza virus proteins analyzed. Considering PB1-F2's role in amplifying inflammation during influenza infection, punicalagin may alleviate inflammation in patients by modulating the function of this protein. Furthermore, the inhibitory effect of punicalagin on PB1-F2 suggests that its anti-influenza activity could also involve inducing structural damage to the viral envelope. This dual mechanism of action underscores punicalagin's potential as a promising therapeutic agent for combating influenza.
