



1. Background
SARS-CoV-2, also known as Severe Acute Respiratory Syndrome Coronavirus-2, is the seventh type of coronavirus. It is an enveloped virus that has a genome consisting of positive-sense, single-stranded RNA (~29.9 kb in size) (1-3). The causative agent of COVID-19 is SARS-CoV-2, which has the potential to become a pandemic (4). The genome of SARS-CoV-2 consists of 11 open reading frames that code for various proteins, including both structural and nonstructural proteins (5-7). Nsp1-16 is essential for viral genome replication and nucleic acid metabolism (8). Additionally, SARS-CoV-2 includes four distinct structural proteins: Membrane (M), spike (S), envelope (E), and nucleocapsid (N) proteins (9). The viral particles have the spike protein on their surface, which interacts with the angiotensin-converting enzyme 2 (ACE2) located on the cell surface. This interaction leads to the internalization of the bound virus into the host cell through endocytosis (10). The spike protein's binding to the ACE2 receptor is what allows viral entry into human target cells (1). Transmembrane protease serine 2, a specific cellular enzyme, activates the spike protein of SARS-CoV-2 by cleaving it into glycosylated subunits called S1 and S2. These subunits play key roles in recognizing receptors and initiating membrane fusion processes (11). The S1 protein can be split into two regions: The N-terminal domain and the C-terminal domain (CTD). The CTD of S1 is particularly important in SARS-CoV-2 as it plays a crucial role in interacting with ACE2 (3).
The residues A475, G476, N487, F465, Y473, Y489, K417, E484, L455, F490, Q493, Y453, Y505, Y449, G496, Q498, T500, N501, G446, and G502 participate in van der Waals (vdW) and hydrogen bond interactions with ACE2 (3). Additionally, spike CTD residue A475 interacts with ACE2 residue S19, N487 with Q24, E484 with K31, and Y453 with H34, exhibiting strong polar interactions (3). Notably, there is a concentration of hydrogen bonds formed between residues G446, Y449, G496, Q498, T500, and G502 of the alpha-1'/beta-1' loop and beta-2'/eta-1' loop in the spike CTD that are located near ACE2 residues D38, Y41, Q42, K353, and D355 (3). Moreover, at the interface of molecules, spike-CTD residues Y489 and F486 interact hydrophobically with ACE2 residues F28, L79, M82, and Y83 by packing together (3). Additional interactions between the virus and receptor involve residue K417, which is situated in helix α3 of the CTD core subdomain. This residue contributes to ionic interactions with ACE2 D30 (3).
Different variants of the SARS-CoV-2 virus have been identified in the United Kingdom (UK) using viral genomic sequencing methods (12). The new variant of COVID-19 has experienced a significant surge in cases in the southeastern region of England (13). According to the sequences of the spike protein in SARS-CoV-2, the monomeric form of the wild-type spike protein includes 1,273 amino acids (NCBI Reference Sequence: YP_009724390.1) (14). This variant, which has 1,270 amino acids and is characterized by multiple spike protein mutations, including deletions at positions 69 - 70 and 144, as well as N501Y, A570D, D614G, P681H, T716I, S982A, and D1118H substitutions, exhibits significantly higher transmissibility than previously circulating variants (13).
2. Objectives
To understand the impact of spike mutations on SARS-CoV-2 and its interaction with ACE2, this study aims to investigate how these mutations affect the structure of the spike protein and its ability to bind to the receptor.
3. Methods
3.1. Structural Analysis
The SARS-CoV-2 spike protein's crystal structure was obtained from the Protein Data Bank with PDB ID 7kj2. This structure is a wild-type prefusion spike glycoprotein that has a single receptor-binding domain positioned to interact with hACE2. A comparison of the sequence of the three-strand spike with the FASTA complete sequence showed that the 7kj2 code had a series of missing residues that needed to be addressed. Additionally, the crystal structure of hACE2 was also obtained from PDB ID 7kj2.
Since the 3D structure of the complete spike protein of SARS-CoV-2 is not available in the PDB, the complete spike protein structure was modeled using MODELLER v9.25 based on homology modeling (15-18). The process of comparative homology modeling involves creating a 3D model by aligning the target sequence, typically represented as a FASTA complete sequence, with a closely related template structure from PDB ID 7kj2 (just the spike protein). This alignment helps generate the most accurate model for the entire sequence. Furthermore, the 3D structure of the UK SARS-CoV-2 spike protein, featuring multiple mutations, including deletion 144, A570D, T716I, D614G, N501Y, P681H, deletion 69-70, S982A, and D1118H, was also modeled using MODELLER v9.25.
3.2. Specifying Residues Involved in Interactions
The interface and critically essential residues involved in interactions between the SARS-CoV-2 spike protein and the ACE2 protein in the 7kj2 structure were examined using LigPlot software (19).
3.3. Molecular Docking
The HADDOCK web server, version 2.4, enables efficient and precise docking of protein complexes through the utilization of biochemical or biophysical data (20, 21). The ranking of single models and clusters is based on the HADDOCK score.
The energy involved in vdW forces between molecules is denoted as Evdw, while the electrostatic energy between molecules is represented by Eelec. Edesol signifies an empirically determined desolvation energy, and Eair corresponds to the AIR energy. To investigate the interaction between the mutated and wild-type SARS-CoV-2-CTD spike proteins with ACE2, the two proteins were docked into the specific sites of the spike protein residues and ACE2 residues using HADDOCK, as inferred from LigPlot.
The results of molecular docking of the mutated spike-ACE2 complex and the completed form of the wild-type spike-ACE2 complex were superimposed using UCSF Chimera 1.15 (22).
3.4. Molecular Dynamic Simulation of Docked Complexes
The molecular dynamics (MD) simulations were conducted using GROMACS 5.2.4 and the GROMOS54a7 force field on an Intel Core i7 Extreme Edition running Ubuntu Linux. For each complex of the mutated spike and wild-type spike-ACE2 complexes, the top-predicted docking pose, specifically the prediction with the first cluster (cluster 1) among all HADDOCK results, was used for the MD simulation. The complex was positioned within a cubic container, ensuring that the solute remained at least 1.0 nm away from any edges of the box. The complexes were solvated with water molecules and simulated using periodic boundary conditions. The system was then neutralized by adding Na⁺ ions and subjected to energy minimization until the maximum forces reached a level below 500 kJ/mol. To maintain a constant temperature, the Berendsen thermostat was utilized throughout all simulations. The pressure of the system was controlled using the Parrinello-Rahman barostat in both the NVT and NPT ensembles. The MD simulation of both the monomeric wild-type and mutated SARS-CoV-2 spike proteins was carried out in 50,000,000 steps (100 ns). To analyze the stability and behavior of the two different forms of the monomeric SARS-CoV-2 spike protein complex with ACE2, a MD simulation was conducted over the course of 50,000,000 steps (100 ns).
3.5. Binding Free Energy Calculation by g-mm-pbsa
The GROMACS molecular mechanics Poisson-Boltzmann surface area (G-MM-PBSA) method was used to calculate the binding free energy of the spike-ACE2 complex during the simulation. In this study, the binding free energies of the spike-ACE2 complex were calculated using the GROMACS tool g_mmpbsa (23-25). The binding free energy of the mutated and wild-type SARS-CoV-2 spike and ACE2 was defined as:
The free energy can be presented as:
4. Results
4.1. Homology Modeling Results
Reference sequence alignment of the spike protein with the NCBI Reference Sequence YP_009724390.1 and the sequence of the PDB file 7kj2 indicated that missing loops and residues are present in these structures. Here, homology modeling was applied using MODELLER v9.25 (http://salilab.org/modeller/) to find the complete 3D structure of the trimeric viral spike protein. The sequence of the spike protein (YP_009724390.1) was used as the primary sequence, and the spike structure in the 7kj2 PDB file was used as the template for MODELLER. Moreover, the structure of the ACE2 protein was extracted from the PDB file of 7kj2.
The structure of the spike protein of SARS-CoV-2 (PDB: 7kj2) was superimposed on the MODELLER model of the complete 3D structure of the trimeric viral spike protein in Chimera using the MatchMaker structural-alignment command tool. The root mean square deviations (RMSD) between 946 pruned atom pairs was 0.567 angstroms (across all 973 pairs: 0.897) (Appendix 1a). These results indicated that some of the turns and loops were deleted in the crystallographic structure of the SARS-CoV-2 spike protein.
To investigate the 3D structure of the UK SARS-CoV-2 spike protein with multiple mutations, including deletion 69 - 70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H, MODELLER v9.25 was used.
The structure of the non-mutated spike of SARS-CoV-2 was superimposed on the MODELLER model of the UK SARS-CoV-2 spike protein with the same multiple mutations in Chimera using the MatchMaker structural-alignment command tool. The RMSD between 1,104 pruned atom pairs was 0.260 angstroms (across all 1,117 pairs: 0.540) (Appendix 1b).
The 3D structure of the wild-type and UK SARS-CoV-2 spike protein with multiple mutations, including deletion 144, A570D, T716I, D614G, N501Y, P681H, deletion 69-70, S982A, and D1118H, is shown using VMD in Appendix 1c.
4.2. Residues Selection
Using LigPlot software, critically essential residues involved in interactions between the SARS-CoV-2 spike protein and the ACE2 protein were examined. Results indicated that residues such as Tyr449, Gly496, Gly502, Asn487, Ala475, Lys417, Thr500, Tyr489, and Phe486 of the SARS-CoV-2 CTD spike interact with residues such as Asp38, Lys353, Tyr41, Gln24, Tyr83, Asp30, Thr27, and His34 of ACE2. The complete results are shown in Appendix 1d.
To investigate the interaction between the mutated and wild-type SARS-CoV-2 CTD spike with ACE2, the two proteins were docked into the specific sites of residues such as Tyr449, Gly496, Gly502, Asn487, Ala475, Lys417, Thr500, Tyr489, and Phe486 of the SARS-CoV-2 CTD spike with residues such as Asp38, Lys353, Tyr41, Gln24, Tyr83, Asp30, Thr27, and His34 of ACE2 using HADDOCK.
Residues involved in interactions between the spike protein and ACE2 protein in the HADDOCK results were examined using LigPlot software (Appendix 1e). The results indicated that the residues involved in spike-ACE2 interaction in the wild-type form of the docked results are very similar to those in the 7kj2 structure. The complete results are shown in Appendix 1e.
4.3. Molecular Dynamics Simulation of Docked Complexes
The MD simulation of two forms of monomeric wild-type and mutated SARS-CoV-2 spike proteins was carried out in 50,000,000 steps (100 ns). To analyze the stability and behavior of the two different forms of the monomeric SARS-CoV-2 spike protein complex with ACE2, a MD simulation was conducted over the course of 50,000,000 steps (100 ns). To compare the two structures, various analyses were conducted. Using LigPlot software, critically essential residues involved in interactions between the SARS-CoV-2 spike protein and ACE2 protein were examined in each 10 ns of the MD simulation results for both the wild-type and mutated SARS-CoV-2 spike-ACE2 complexes (Appendix 2). The results indicated that hydrogen bonds, hydrophobic interactions, and double/triple bonds were more prevalent in the mutated spike-ACE2 complex.
4.4. Root Mean Square Deviations
An important parameter for assessing the conformational stability of proteins and protein-protein complexes in a dynamic state during simulation is the RMSDs between C-α atoms on the protein backbone. RMSDs were calculated by comparing each frame to the initial conformation, and plots of RMSD against simulation time were used to analyze overall system behavior. Consistent fluctuations with low levels of RMSD throughout the simulation suggest that the system has reached equilibrium and stabilization. Conversely, higher fluctuations in RMSD indicate lower stability of the system. Additionally, significant deviations in RMSD graphs may suggest major conformational changes occurring within the protein as it attempts to achieve a stable configuration with its receptor site.
The RMSDs for the wild-type spike protein backbone Cα atoms, the mutant spike protein backbone Cα atoms, and the wild-type spike- and mutant spike-ACE2 complexes were calculated and produced as a graph depicted in Appendix 3. Appendix 3 revealed significant discrepancies in the RMSD observed between the wild-type monomeric spike protein and its mutated counterpart (a), the wild-type spike-ACE2 complex and the mutant spike-ACE2 complex (b), ACE2 in the wild-type spike-ACE2 complex and ACE2 in the mutant spike-ACE2 complex (c), and the active site of the wild-type spike and the active site of the mutant spike in the spike-ACE2 complex (d).
The RMSD value of the monomeric spike protein with multiple mutations, including deletion 69-70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H, increased gradually from 20 ns to 100 ns. This suggests that there was evidence of instability in the protein due to mutations in its spike. In contrast, the graph showed consistent and stable patterns without significant fluctuations observed across different time intervals among the trajectories. These results imply that the protein backbone underwent major structural perturbations in the mutant form of the spike. The RMSD of the wild-type monomeric spike model increased suddenly and stabilized at 80 ns. Fluctuations were observed during the simulation and then suddenly increased at 32, 60, 82, and 100 ns to 2.4, 2.7, 2.9, and 3.2 nm in the mutant form of the spike protein, whereas for the wild-type monomeric spike, fluctuations were observed at 31 ns and 45 ns. Based on these findings, it can be concluded that the wild-type monomeric spike protein maintained a consistent conformation throughout the simulation, with only minimal changes in structure occurring (Appendix 3a).
The root mean square deviations were computed for the protein backbone Cα atoms of both the wild-type spike-ACE2 complex and the mutant spike-ACE2 complex, which includes multiple mutations such as deletion 69-70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H in the spike protein. These calculations resulted in a graph shown in Appendix 3b, illustrating the range of RMSD values obtained. The magnitude of these RMSDs varied greatly, with a minimum value of 0.0000047 nm and a maximum value of 3.29 nm when comparing the interactions between the wild-type spike and ACE2 versus the mutant spike and ACE2, respectively.
In the complexes of wild-type spike-ACE2 and mutant spike-ACE2, the data showed stable trajectories with small fluctuations. This suggests that there were minor changes in the structural configurations of these complexes. The mutant spike-ACE2 complex exhibited lower RMSD values after 10 ns until 100 ns compared to the wild-type spike-ACE2 complex. This was a strong indication of conformational stability in the mutant spike with deletions and mutations in complex with the ACE2 ligand, relative to the wild-type spike-ACE2.
Moreover, noticeable differences in the paths taken by the wild-type spike-ACE2 and mutant spike-ACE2 complexes were observed at different time intervals. Specifically, after 10 ns, the RMSD fluctuation of the wild-type and mutant spike-ACE2 complexes became much more prominent, indicating substantial conformational and structural changes. The variations in movement were initially detected at 10 ns, with the RMSD of the two complexes demonstrating a gradual increase for the wild-type spike-ACE2 and a subsequent decrease for the mutant spike-ACE2. The fluctuations ranged from 1.1 nm upwards and gradually decreased accordingly.
The experimental findings showed that the spike-ACE2 complex with the mutations of deletion 69-70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H maintained a consistent conformation throughout the simulation, experiencing minimal changes in structure. Appendix 3c illustrates the range of RMSD values for ACE2 in both the wild-type spike-ACE2 complex and the mutant spike-ACE2 complex, indicating that there were stable trajectories with small fluctuations.
The RMSD of the backbone Cα atoms for the active site of the wild-type spike and the active site of the mutant spike in the spike-ACE2 complex indicated that the variations in movement were initially detected at 30 ns. The RMSD values of the two complexes demonstrated a gradual increase for the active site of the mutant spike in the spike-ACE2 complex and a subsequent decrease for the active site of the wild-type spike in the spike-ACE2 complex (Appendix 3d).
4.5. Root Mean Square Fluctuation
Every individual amino acid in the protein is crucial for attaining a stable structure of both the protein itself and the receptor-ligand complex. The extent of stability can be assessed by analyzing the root mean square fluctuation (RMSF) of the Cα atom. By plotting RMSF against residue number, we can determine the average fluctuation experienced by each amino acid throughout the MD simulation.
The graph of RMSF shows the fluctuation values on the y-axis against the residues depicted on the x-axis. The plot demonstrates different levels of flexibility for each residue in the spike protein. Higher RMSF fluctuations indicate increased flexibility, suggesting a greater potential for interaction. Similarly, residues of the spike protein with low flexibility indicate their decreased potential to interact with the ACE2 receptor molecule. The RMSFs for the residues of both wild-type and mutant spike proteins, as well as the wild-type spike-ACE2 complex and the mutant spike-ACE2 complex, were computed. These calculations yielded a graph presented in Appendix 4, depicting notable fluctuations in flexible residues with prominent peaks.
For the monomeric wild-type spike and mutant spike protein with multiple mutations, including deletion 69 - 70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H, it is evident that the fluctuation of the mutant spike protein structure is greater compared to the wild-type spike structure in the RMSF graph during the MD simulation. The protein residues involved in ligand binding, specifically those ranging from residues 370 to 510 in the mutant spike, exhibit higher RMSF values, implying greater fluctuation in these protein regions.
In the mutant spike protein, fluctuations were observed in regions from residue indices 282 - 484, 500 - 540, 547 - 606, 608 - 692, 708 - 733, 756 - 778, 849 - 898, 934 - 977, 996 - 1024, and 1037 - 1117. The highest fluctuations were noted between residues 313 - 325, 409 - 424, 443 - 480, 501 - 520, 667 - 689, 939 - 967, and 1044 - 1117, suggesting these regions could be affected by multiple mutations. Specific residues such as THR316, ASP376, VAL416, GLN469, and LYS1057 in the mutant spike protein displayed pronounced fluctuations. In contrast, the wild-type spike exhibited more fluctuations in residues SER433 and LEU815. It is clear that there is a higher fluctuation in the mutant spike protein compared to the wild-type spike. In other words, as observed from the RMSF plot, the mutant spike protein structure is less stable compared to the wild-type spike structure (Appendix 4a).
Appendix 4b illustrates the analysis of RMSF trajectories for the spike in both the wild-type ACE2 complex and the mutant spike-ACE2 complex. In the case of the spike-ACE2 complex, the RMSF analysis revealed that certain residues of the spike protein deviated from their average structure upon interaction with ACE2 receptors. In the wild-type spike-ACE2 complex, fluctuations were observed in regions from residue indices 313-564 in the wild-type spike protein. These fluctuations imply that these residues could be involved in protein-receptor binding. On the other hand, in the mutant spike-ACE2 complex, fluctuations were low in the RMSF plot for the spike protein with multiple mutations including deletion 69-70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H.
Additionally, the RMSF of the backbone Cα atoms of ACE2 in both the wild-type spike-ACE2 complex and the mutant spike-ACE2 complex are shown in Appendix 4c. In the case of the mutant spike-ACE2 complex, fluctuations were also low in the RMSF plot for ACE2. These results indicate that the mutant spike-ACE2 complex is more stable than the wild-type spike-ACE2 complex.
4.6. Radius of Gyration
In order to examine the impact of multiple mutations (deletion 69 - 70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H) on the compactness of spike proteins and their complex with the ACE2 receptor, we conducted calculations for the radius of gyration (Rg). Radius of gyration is a metric that indicates the distance between atoms in a system relative to its center or axis. By plotting Rg over simulation time, it provides insights into the compactness of protein structures influenced by various combinations of secondary structures. The stability of protein folding can be inferred from a relatively consistent Rg value throughout; however, in unfolded proteins, the Rg will change over time.
In addition to the analysis of the compactness and folding of the unbound protein, the Rg parameter was also used to analyze the compactness of the protein-ligand complexes. Weak intermolecular bonds promote a loss of compactness in the system and affect the stability of the complex. When the interactions between the ligand and protein are weaker, the compactness of the protein-ligand complex decreases, resulting in a higher Rg value for the complex.
Figure 1A illustrates how the Rg of the mutant monomeric spike changes over time compared to the wild-type monomeric spike. The graph depicting the Rg values over time clearly demonstrates that the mutant monomeric spike consistently remains in an unstable compact (unfolded) state throughout the 100 ns duration. This suggests that the protein's Rg is highly volatile in the case of the mutant variant. The Rg value in the complex ranges between 3.8 and 4.9 nm, and it gradually balances at 55 ns.
The Rg trajectories of the wild-type and mutant spike-ACE2 complexes were also investigated. As shown in Figure 1B, it is evident that the mutant spike-ACE2 complex is more compact compared to the wild-type spike-ACE2 complex. From the beginning of the simulation process, it can be concluded from Figure 1 that the mutant spike-ACE2 complex has lesser compactness compared to the wild type, and at the end of the simulation, higher fluctuations indicate a loss of compactness.
 of the backbone Cα atoms of wild-type spike and mutant spike protein (A) and the wild-type spike-ACE2 complex and mutant spike-angiotensin-converting enzyme 2 (ACE2) complex (B).")
Radius of gyration (Rg) of the backbone Cα atoms of wild-type spike and mutant spike protein (A) and the wild-type spike-ACE2 complex and mutant spike-angiotensin-converting enzyme 2 (ACE2) complex (B).
4.7. Solvent Accessible Surface Area Analysis
The solvation free energy of a protein is determined by the way polar and non-polar residues interact with each other. The solvent molecule's probe monitors the surface area of the protein to find and assess the vdW surface of the protein. The solvent-accessible surface area (SASA) has been used for predicting changes in solvent-accessible area induced by residue changes or ligand interactions. Among all residues, hydrophobic residues are primarily responsible for increasing the SASA value. The residue SASA value, an important parameter in SASA analysis, provides an understanding of protein conformational changes per residue contribution. Indeed, the interaction of the protein and protein-ligand complex with various solvents, or the interaction of the protein alone with both various solvents and ligands, primarily depends on their surface properties.
The last 100 ns trajectory was used for the calculation of the SASA value, as plotted in Figure 2. Results indicated that the values fluctuate more in the mutant spike protein and wild-type spike-ACE2 complex than in the wild-type spike protein and mutant spike-ACE2 complex, respectively, indicating greater compactness of the latter. Hence, we analyzed the average SASA value for all the systems. The average SASA values for the wild-type spike protein, mutant spike protein, wild-type spike-ACE2 complex, and mutant spike-ACE2 complex were 562.09, 564.39, 827.19, and 821.78 nm², respectively. The findings from this study suggest that the simultaneous occurrence of multiple mutations, including deletions at positions 69 - 70 and 144, along with N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H on the spike protein, has a minimal impact on the exposed surface area of its structure.
 complex and mutant spike-ACE2 complex.")
The SASA of the four structures wild-type spike protein, mutant spike protein, wild-type spike- angiotensin-converting enzyme 2 (ACE2) complex and mutant spike-ACE2 complex.
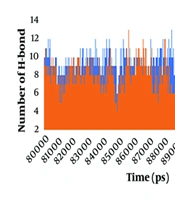
4.8. Hydrogen Bond Analysis
The formation of hydrogen bonds between the ligand and the protein is a crucial factor in substrate recognition and in maintaining the stability of the ligand-protein complex. A hydrogen bond occurs when there is an angle of less than 30° between the hydrogen donor and acceptor, with a distance shorter than 0.3500 nm. To enhance our visual analysis of the hydrogen bonding between the spike protein and ACE2 protein during the simulation, we examined the number of observed hydrogen bonds, as depicted in Figure 3. The number of hydrogen bonds formed between the spike protein and the ACE2 protein was analyzed in both the wild-type and mutant spike-ACE2 complexes. The results show that during the simulation period of 8000-9500 ns, the wild-type complex exhibited a range of 4 to 13 hydrogen bonds, with an average of approximately 8 hydrogen bonds. On the other hand, in the mutant complex, at least 3 hydrogen bonds were observed, with a maximum of 13 bonds. The average number of hydrogen bonds decreased to approximately 7 during the 8000 - 9500 ns simulation in the mutant spike-ACE2 complex. The hydrogen bond formation between the spike and ACE2 proteins emphasized that they had a strong and stable binding before the multiple mutations of deletion 69-70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H occurred in the spike protein.
 in wild-type spike-ACE2 complex and mutant spike-ACE2 complex.")
H-bond formed by spike and angiotensin-converting enzyme 2 (ACE2) in wild-type spike-ACE2 complex and mutant spike-ACE2 complex.
4.9. Principal Component Analysis
Using principal component analysis (PCA), an analysis of eigenvectors and eigenvalues was performed. In MD simulations, PCA obtains the associated motion of the protein and protein-ligand complex to define the protein's function. In the calculation of the principal components (PCs) or essential dynamics (ED), the conformational space of the protein, which contains a few degrees of freedom despite the protein's motion, was identified.
The total movement in the system is equal to the combined values of the eigenvalues, making it a useful tool for assessing the protein's flexibility under various conditions. GROMACS offers a feature that allows for calculating eigenvectors to analyze protein motions before and after applying multiple mutations (deletion 69-70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H) in the spike protein-ACE2 complex. The principal motion was used to characterize protein motion by eigenvector and eigenvalues. Hence, in this study, PCA was performed to analyze the atomic motions of wild-type spike protein, mutant spike protein, wild-type spike-ACE2 complex, and mutant spike-ACE2 complex using Cartesian coordinates of the Cα atoms. We plotted the eigenvalues with the eigenvectors for the 1500 eigenvectors during the 8000 - 9500 ns of the simulation in Figure 4.
 of wild type monomeric spike (A), mutated monomeric spike (B), wild type spike- angiotensin-converting enzyme 2 (ACE2) compelexes (C) and mutated spike-ACE2 complexes (D).")
The principal component analysis (PCA) of wild type monomeric spike (A), mutated monomeric spike (B), wild type spike- angiotensin-converting enzyme 2 (ACE2) compelexes (C) and mutated spike-ACE2 complexes (D).
Due to the clear depiction of the calculation of eigenvalue vs. eigenvector, the result was plotted in Figure 4A, selecting only the first 40 eigenvectors for wild-type spike protein, mutant spike protein, wild-type spike-ACE2 complex, and mutant spike-ACE2 complex. The mutant spike-ACE2 complex showed very high motion compared to the wild-type spike-ACE2 complex, while the wild-type spike protein and mutant spike protein exhibited significantly less motion. This indicates that the multiple mutations—deletion 69 - 70, deletion 144, N501Y, A570D, T716I, D614G, P681H, S982A, and D1118H—in the spike-ACE2 complex induce structural changes and motions in the protein. Furthermore, both the wild-type spike-ACE2 complex and mutant spike-ACE2 complex demonstrated substantial motions compared to the wild-type spike protein and mutant spike protein.
To clearly understand the motions induced by the multiple mutations, the correlated motions (averaged) were calculated for 1500 eigenvectors during the 8000 - 9500 ns of the simulation. The wild-type spike protein, mutant spike protein, wild-type spike-ACE2 complex, and mutant spike-ACE2 complex showed 4%, 3%, 11%, and 8% correlated motions, respectively.
4.10. Binding Free Energy Calculation by g-mm-pbsa
The GROMACS molecular mechanics Poisson-Boltzmann surface area (G-MM-PBSA) method was used to calculate the binding free energy of the wild-type spike-ACE2 complex and the mutant spike-ACE2 complex during the simulation. The results are shown in Table 1. The highest interaction energy was associated with the mutant spike-ACE2 complex.
| Variables | VdW (kj/mol) | Elec (kj/mol) | Total (kj/mol) | Hbond |
|---|---|---|---|---|
| Wild-type spike-ACE2 complex | -277.49 | -893.97 | -1171.47 | 8.37 |
| Mutant spike-ACE2 complex | -300.98 | -1352.22 | -1653.20 | 7.38 |
The Mean Interaction Energies and Hydrogen Bonding of Wild-Type Spike- Angiotensin-Converting Enzyme 2 Complex and Mutant Spike-ACE2 Complex
5. Discussion
A SARS-CoV-2 variant has been identified in the UK, which is significantly more transmissible than previously circulating variants. The effects of UK SARS-CoV-2 mutations on spike functions and its interactions with ACE2 were studied using molecular dynamic simulations, employing RMSD, RMSF, RG, PCA, and SASA h-bond commands in Gromacs 5.2.4. Root mean square deviations analysis indicated that the wild-type monomeric spike protein achieved a stable conformation during the simulation, while the mutant spike-ACE2 complex also attained a stable conformation, albeit with a few conformational transitions.
Analysis of the RMSF plot indicated that the mutant spike protein structure is less stable compared to the wild-type spike structure. Radius of gyration values demonstrated that the mutant monomeric spike maintains a very unstable compact form, indicating that the RG of the protein is quite unstable in the mutant protein. Additionally, the RG trajectories of the wild-type and mutant spike-ACE2 complexes showed that the mutant spike-ACE2 complex is more compact compared to the wild-type spike-ACE2 complex, although the mutant spike-ACE2 complex exhibited less compactness compared to the wild type, with higher fluctuations at the end of the simulation indicating a loss of compactness.
Solvent accessible surface area analysis revealed that the values fluctuate more in the mutant spike protein than in the wild-type spike protein, and in the wild-type spike-ACE2 complex compared to the mutant spike-ACE2 complex. Furthermore, the number of hydrogen bonds formed between the spike protein and the ACE2 protein in the wild-type spike-ACE2 complex is greater than that in the mutant spike-ACE2 complex. Principal component analysis indicated that the mutant spike-ACE2 complex exhibited significantly higher motion compared to the wild-type spike-ACE2 complex. The G-MM-PBSA method indicated that the highest interaction energy was associated with the mutant spike-ACE2 complex. Moreover, both the wild-type spike protein and mutant spike protein displayed minimal motion, while the PCA analysis of the wild-type spike-ACE2 complex and mutant spike-ACE2 complex showed very high motions compared to the wild-type spike protein and mutant spike protein.
