Several studies have shown that degenerative mechanisms may be due to mitochondrial dysfunction (
27). Related phenomena, such as radical changes, have been elucidated in MS pathogenesis (
28), and reduction of COX gene expression in the blood of MS patients was observed (
29). One of the important results of the present study is the significant reduction of ATP value in the EAE group, compared to the negative and sham controls. The lowering ATP can be attributed to the low specific activity of COX, which is determined and confirmed. In 2010, proteome analysis of the EAE mice brain revealed a significant reduction of expression of two subunits of the COX enzyme (Cox5b, Cox5a) involved in the structure and accumulation of complex IV of mitochondria (
17). The authors concluded that low energy in EAE mice's brains is similar to the hypoxia-like damage that occurred in the CNS of the MS patients. These conditions could be characterized by a gradual increase of the HIF-1α and accumulate in hypoxic conditions in associating with HIF-1β to form a functional complex, triggering to do transcription a large collection of hypoxia-inducible genes involved in nerve protection leading to better adaption, and survival of cells exposed to hypoxia. HIF-1α may increase blood oxygen, glucose supply, compensate for the reduced ATP supply with glycolytic pathways by regulating their genes (
30). In the case of animal models, mice deficient for HIF-1 showed significantly less neuronal cell loss than control mice in response to hypoxia (
31). Plenty of evidence indicated that potential neuroprotective roles of HIF-1α are involved in the pathogenesis of Alzheimer’s, stroke, and inflammatory brain diseases (
32-
35). In spite of the several reports about the activation/inactivation of HIF-1α in cell-mediated inflammation, the specific role of HIF-1α in the pathogenesis of the disease is still unknown (
36). However, autopsy and microarray studies on the MS patients indicated that mitochondrial damage and related hypoxia-like conditions in white matter are the important pathway of tissue injury in MS disease. Oxidative damage, ROS, and NO has been implicated and used in the explanation of hypoxia-like observations (
37,
38). One of the important reports is the actual hypoxia in the white matter of the CNS of the EAE rat model presented by Davies in 2013. They tried to assess the brain hypoxia in vivo and reducing the hypoxia-like condition by administration of brief and continues normobaric oxygen (
18). They also tried to administrate the iNOS inhibitors by borrowing the method of Zielasek et al., to improve the hypoxia-like condition, as well (
39). They concluded that the neurological deficit was closely correlated with spinal white and gray matter hypoxia. Consistent with this finding, we planned to assess the HIF-1α, ATP, and specific activity of COX enzyme in whole-brains of the EAE mice model of MS. The data also showed that HIF-1α was significantly increased in the EAE mice group, compared to the negative control and sham groups. The only report that is not relatively consistent with these data is the published paper by Moan (2015) (
40), who described in spite of the significant increase of the HIF-1α in astrocytes and myeloid in spinal cord samples of the EAE mice, the expression level of this factor in those cells was not necessary for the development of the neuro-inflammatory disease. However, there are important reports which showed that induction of HIF-1α may be influential, as the neuroprotective agent for other neurodegenerative disorders such as Alzheimer's disease (
32) or as a therapeutic target in the ischemic stroke (
34). In 2014, Li et al. described that up-regulated HIF-1α may be involved in the process of epileptogenesis but not in the acute stage of epilepsy in animal models and claimed that the modulation of HIF-1a may offer a novel therapeutic target in epilepsy (
41). In 2018, Navarrete et al. tried to use a special compound (VCE-004.8) to mimic hypoxic conditions by stabilizing the HIF-1α and HIF-2α as well as activating the HIF pathway in different cell types of two animal models of MS. In vivo experiments of this study revealed that the VCE-004.8 treatments could prevent demyelination, axonal damage, and immune cell infiltration (
42). It seems the HIF-1α induction may also be a potential target to control MS progression.
In conclusions, the present study, for the first time, showed that in peak score of MS disease in the EAE mouse model, specific COX activity and, consequently, ATP levels of the mice brain cells were significantly decreased, indicating the presence of hypoxia-like conditions. Significant induction of HIF-1α in the EAE mice brains also implies indirectly hypoxia-like conditions. This indicates that enhanced HIF-1α may compensate for reduced ATP supply resulted from the COX activity loss in compromised mitochondria and could prevent neuronal death under hypoxia-like conditions in the EAE mice brains. In general, it seems that these data may help to elaborate on the role of HIF-1α as the neuroprotective agent for MS disease.
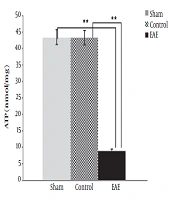


 in whole brain of each group of experimental mice. Black column = EAE mice group, Dark gray = Negative control of mice group, and Gray column = Sham group. ** Statistically significant with a P value less than 0.005")