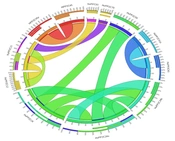
4.2. Identification of the Key Module Correlated With Salt Stress in the Glucose-TOR Pathway
Next, we examined the correlations between module eigengenes and sample traits to identify salt-responsive modules. This analysis aimed to determine which module responded consistently to salt stress across different time points.
The results showed variable expression patterns among glucose-TOR downstream genes under salt stress (
Figure 2). The turquoise and green modules showed strong positive correlations with salt treatment during the early stress phase (3 hours), suggesting rapid pathway activation. The brown module showed the highest correlation under prolonged treatment (7 days). The green module showed the strongest positive correlation with salt stress. The turquoise module showed transient early activity, whereas the yellow module was strongly negatively correlated, indicating downregulation. In addition, the green module showed only a weak negative correlation with time (r = -0.49), suggesting initial activation with sustained expression. Its low correlation with biological replicates (r = +0.08) also indicates robustness and low random noise (
Table 2).
| Module | Time | Salt Treatment | Replicates |
|---|
| Blue | -0.94729 | -0.09868 | 0.16443 |
| Brown | 0.979922 | 0.029209 | 0.174907 |
| Green | -0.49409 | 0.7575466 | -0.08272 |
| Grey | -0.077 | 0.237394 | -0.07557 |
| Red | 0.648724 | -0.21676 | -0.055409 |
| Turquoise | -0.32089 | 0.421813 | -0.06582 |
| Yellow | 0.369367 | -0.79874 | 0.01352 |
Heatmap of correlations between gene expression modules and experimental samples. Blue indicates positive correlation, and red indicates negative correlation.
Given its strong and consistent correlation with salt stress, low variability, and sustained expression, the green module was selected as the key TOR-associated module in the salt-stress response.
4.4. Functional Roles of Hub Genes in the Green Module Suggest Adaptive Strategies
To identify central genes in the green module, co-expression networks were constructed in Cytoscape, and kME values were used to define hub genes. Genes with kME > 0.96 were selected, yielding 6 hub genes. Their positions in the network, particularly their connections to other genes, are shown in
Figure 4. These genes not only occupied central topological positions but also showed strong interconnectivity, highlighting their regulatory significance under salt stress.
Co-expression network of green-module genes in S. parvula. Nodes represent genes; thicker edges denote stronger co-expression. Dark green nodes indicate higher kME values, and hub genes are shown on the left.
Subsequent analysis of hub-gene topology and expression supported their central roles in the stress response (
Table 4). Most hub genes were significantly upregulated under NaCl treatment at various developmental stages (
Figure 5). Functional annotations and ortholog comparisons with
A. thaliana confirmed their involvement in cell division, detoxification, root growth, and stress response.
| Gene Code | kME | Degree | Mean Edge Weight | Arabidopsis Ortholog | Gene Annotation | MapMan Ontology | Gene Function | Reference(s) |
|---|
| Sp4g25020 | 0.9812 | 62 | 0.3849 | AT2G42890 | MEI2-like protein | RNA.RNA binding | Meiosis, cell division, vegetative growth, response to ABA | (13) |
| Sp1g40550 | 0.9732 | 66 | 0.3946 | AT1G54100 | Aldehyde dehydrogenase | Fermentation. Lactate dehydrogenase | Cellular detoxification, adaptation, salt tolerance, root growth | (16) |
| Sp4g20000 | 0.9702 | 62 | 0.3809 | AT2G37640 | Alpha-expansin family | Cell wall. Modification | Cell wall loosening for division and elongation | (14) |
| Sp4g20790 | 0.9662 | 66 | 0.3754 | AT2G38465 | Uncharacterized | Not assigned. Unknown | Unknown | - |
| Sp1g05330 | 0.9627 | 68 | 0.3874 | AT1G06570 | 4-hydroxyphenylpyruvate dioxygenase | Secondary metabolism. Isoprenoids. Tocopherol | Biosynthesis of plastoquinone and tocopherols, abiotic stress tolerance | (20) |
| Sp4g22080 | 0.9624 | 62 | 0.3558 | AT2G39800 | delta1-pyrroline-5-carboxylate synthase (P5CS) | Amino acid metabolism. Synthesis. Glutamate family. Proline | Proline biosynthesis, salt-stress tolerance | (17) |
Volcano plots showing expression of green-module hub genes at A, 3 hours; B, 24 hours; C, 48 hours; D, 3 days; and E, 7 days after NaCl treatment. Blue indicates upregulation, pink indicates downregulation (P < 0.05), and grey indicates no significant change.
Among the hub genes in the green module, Sp4g25020 had the highest kME value and exhibited a high degree of connectivity in the network (kME = 0.9812; degree = 62). This gene is the ortholog of AT2G42890 in
A. thaliana, which encodes a protein from the MEI2-like family. This family is known to regulate meiotic and mitotic cell divisions and vegetative growth, and it also plays a role in the response to ABA (
13). These characteristics underscore the importance of Sp4g25020 as a central regulator in salinity-stress adaptation.
Another gene involved in cell division is Sp4g20000, which was significantly upregulated in the roots of
S. parvula under all salt-stress treatments (P < 0.01;
Figure 5). The ortholog of this gene in
A. thaliana belongs to the alpha-expansin family, which plays a key role in loosening the cell wall during cell division and elongation. Members of this family are expressed in the root growth zone, and their downregulation leads to reduced root growth (
14). Increased expression of expansin genes in response to abiotic stresses in roots has also been reported (
15). Therefore, the high expression of this gene under salinity may be associated with the structural adaptation of roots to stress conditions.
Another hub gene with high kME and degree values is Sp1g40550. This gene had the highest mean edge weight among the hub genes (mean edge weight = 0.3946), indicating stronger connectivity with co-expressed genes and a more influential role within the co-expression network. It encodes an aldehyde dehydrogenase that reduces toxic aldehyde levels by oxidizing them. Overexpression of this gene in
A. thaliana has been shown to enhance tolerance to abiotic stresses, including salinity, whereas its silencing increases sensitivity to NaCl and inhibits root growth (
16). The presence of this gene in the co-expression network may indicate the importance of detoxification and free-radical control in response to oxidative stress under salt conditions.
Another important hub gene in the green module is Sp4g22080. This gene was significantly upregulated under all salt treatments compared with the control (P < 0.01;
Figure 5). Its ortholog in
A. thaliana encodes delta1-pyrroline-5-carboxylate synthase (P5CS), a key and rate-limiting enzyme in the proline biosynthesis pathway. P5CS expression increases in roots under NaCl treatment (
17). Proline functions as a major osmolyte in response to salinity, and its accumulation is associated with plant stress tolerance (
18). Independent of plant hormones, this amino acid regulates cell division in the meristematic zone of
A. thaliana roots and determines meristem size by modulating division-to-differentiation rates (
19).
Sp1g05330 catalyzes the first step in the biosynthetic pathway of tocopherol (vitamin E) and plastoquinone, both of which are crucial for antioxidant protection in plants. Overexpression of the ortholog of this gene in sweet potato has been associated with increased tolerance to salt, drought, and oxidative stresses (
20). Under salinity, preventing oxidative stress in roots is vital for maintaining structural and physiological integrity.
Among the hub genes, Sp4g20790 does not have a clearly defined function. However, its prominent network position (kME = 0.9662; degree = 66) suggests an important role that remains experimentally uncharacterized. Such genes are valuable targets for future functional studies.
These findings suggest that S. parvula employs a complex TOR-regulated network involving the coordinated control of cell division and expansion, detoxification, osmoprotectant synthesis, and antioxidant defense to maintain root growth and stress tolerance under salinity.





.")

, and grey indicates no significant change.")