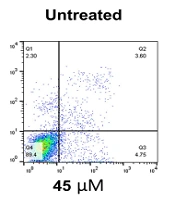
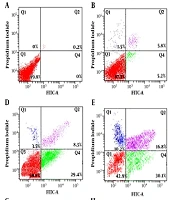
Cervical cancer, as a common malignancy, is associated with substantial mortality and morbidity (
16). Key therapeutic modalities include surgery, radiotherapy, chemotherapy, and immunotherapy; each is linked to notable adverse effects (
17), which necessitates the development of new therapeutic approaches. In this research, we evaluated the impacts of varying concentrations of EGCG on two cervical cancer cell lines, HeLa and CaSki. Our findings show that EGCG exerts cytotoxic effects, particularly at higher concentrations (40 and 80 μg/mL), against cancer cells, and cancer cell death was accompanied by apoptosis. Gene expression analysis revealed that EGCG upregulated
bax,
CASP3,
CASP9, and
TP53 (
p53) transcripts, while downregulating
bcl-2 and
EGFR transcripts in cancer cells. Collectively, these results suggest that the cytotoxic effects of EGCG are mediated, at least in part, by activation of the mitochondrial (intrinsic) apoptotic pathway and by inhibition of proliferative signaling via the EGFR pathway.
The EGCG is one of the most important catechin compounds in several plants, particularly green tea, and has shown a broad spectrum of biological effects, including antioxidant, anti-obesity, anti-inflammatory, anti-diabetic, and anticancer effects (
18). The anticancer activities of this natural product are exerted through modulation of multiple signaling pathways (
12). In this study, we observed that EGCG exerted anticancer effects against both HeLa and CaSki cervical cancer cell lines, accompanied by the induction of apoptosis. Our findings align with numerous studies reporting anticancer effects of EGCG across a variety of malignancies (
19-
21). Notably, in this study, 40 μg/mL EGCG had strong cytotoxic effects against both ovarian cancer cell lines. This contrasts with Zhang et al., who reported that 80 μg/mL EGCG induced maximal cell death in hepatocellular carcinoma HCCLM6 cells, suggesting cell-type–specific sensitivity to EGCG (
22). Differences in cell type may account for this discrepancy, indicating that HeLa and CaSki cells may be more sensitive to EGCG. Another study reported that 40 μg/mL EGCG significantly inhibited proliferation of Hep3B cells (
23), consistent with our observation that this concentration can induce substantial cytotoxic effects.
This study showed that EGCG overexpressed
bax,
CASP3,
CASP9, and
p53 genes, while downregulating
bcl-2 and
EGFR in cancer cells. These findings indicated that EGCG promotes a shift toward pro-apoptotic signaling in cancer cells, as evidenced by the upregulation of
bax,
CASP3,
CASP9, and
p53 transcripts alongside the downregulation of anti-apoptotic
bcl-2 and the growth/survival receptor
EGFR. Increased
bax can disrupt mitochondrial membrane integrity, facilitating cytochrome c release and activation of the caspase cascade, highlighted by elevated
CASP9 and
CASP3 transcriptional signals (
24). The concurrent rise in
p53 suggests a p53-mediated transcriptional response to cellular stress, reinforcing apoptotic pathways and potentially enhancing DNA damage surveillance (
25,
26). Suppression of
bcl-2 removes a critical brake on mitochondrial outer membrane permeabilization, synergizing with Bax to promote apoptosis (
27). Reduced EGFR transcripts may diminish proliferative and survival signaling, further tipping the balance toward cell death rather than growth (
28,
29). Collectively, these gene expression changes support a mechanistic model where EGCG activates intrinsic apoptosis and suppresses pro-survival cues, offering a molecular rationale for its anticancer effects in this context.
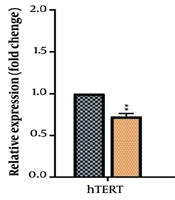
It seems that EGCG elicits a dose-dependent alteration of key regulatory pathways in cervical cancer cells, upregulating the tumor suppressor p53 while concurrently suppressing the receptor tyrosine kinase EGFR. The concordant changes in p53 and EGFR expression may reflect a coordinated cellular response to EGCG that contributes to reduced proliferation and increased apoptotic signaling in these cell lines. However, this study was conducted in vitro, which is one of the limitations of this study, and generalization of the findings of this research to clinical conditions should be done with caution. Therefore, further mechanistic studies are warranted to dissect the upstream regulators mediating EGCG’s effects on p53 and EGFR expression. Finally, functional validation (protein levels) would strengthen the link between transcript changes and cell fate.
5.1. Conclusions
Our data indicate that EGCG exerts dose-dependent cytotoxic effects on cancer cells, with the most pronounced cell death observed at higher concentrations (40 and 80 μg/mL), predominantly via apoptosis. The observed transcriptional profile supports activation of the intrinsic (mitochondrial) apoptotic pathway, evidenced by upregulation of pro-apoptotic bax, caspases (CASP3 and CASP9), and p53, together with downregulation of anti-apoptotic bcl-2. Concurrent suppression of EGFR suggests reduced pro-survival signaling, which may further sensitize cells to apoptotic death. Collectively, these results propose a mechanistic model in which EGCG initiates mitochondrial-dependent apoptosis and attenuates growth/survival pathways in cancer cells. Future studies validating these findings at the protein level and assessing functional apoptosis will strengthen the translational potential of EGCG as an anticancer agent.
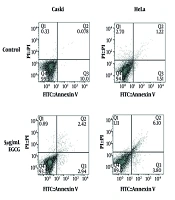





 and the apoptosis rate (B) after treatment with 0, 5, 10, 20, 40, and 80 µg/mL epigallocatechin-3-gallate (EGCG). Cells were treated with EGCG for 72 hours, and cell viability and apoptosis were measured by MTT assay and flow cytometry, respectively (n = 3).")

 and <i>bcl-2</i> (B) genes in HeLa and CaSki cell lines after treatment with 0, 5, 10, 20, 40, and 80 µg/mL epigallocatechin-3-gallate (EGCG). Cells were treated with EGCG for 72 hours, and gene expression was measured by qRT-PCR (n = 3).")
 and <i>CASP9</i> (B) genes in HeLa and CaSki cell lines after treatment with 0, 5, 10, 20, 40, and 80 µg/mL epigallocatechin-3-gallate (EGCG). Cells were treated with EGCG for 72 hours, and gene expression was measured by qRT-PCR (n = 3).")
 and <i>EGFR</i> (B) genes in HeLa and CaSki cell lines after treatment with 0, 5, 10, 20, 40, and 80 µg/mL epigallocatechin-3-gallate (EGCG). Cells were treated with EGCG for 72 hours, and gene expression was measured by qRT-PCR (n = 3).")