Chemicals
Aβ1-42, 4-2-hydroxyethyl-1-piperazineethanesulfonic acid (
HEPES), rotenone (
Rot), dimethyl sulfoxide (
DMSO), D-mannitol, thiobarbutiric-acid (
TBA), 2′,7′-Dichlorofluorescin diacetate (
DCFH-DA), Tris–HCl, sodium-succinate, phosphate-buffered saline (
PBS), sucrose, KCl, Na
2HPO
4, MgCl
2, Rhodamine123 (
Rh123), Coomassieblue, ethylene glycol bis (2-aminoethyl ether)-N, N, N0, N0-tetraacetic acid (
EGTA), and xylazine and ketamine were purchased from Sigma Chemical Co. (St. Louis, MO, USA). All chemicals used for this experiment were of the best analytical and pharmaceutical grade available. Crocin was quantified through the original method in an aqueous saffron extract (
27). Crocin was 95% pure. Aβ peptide1-42 was dissolved in PBS at a concentration of 20 µg/µL, aliquoted and stored at -80 ˚C until use (
40).
Animals
Male rats of the Wistar strain weighing 220–250 g were obtained from the Faculty of Pharmacy, Shahid Beheshti University of Medical Sciences. All animal groups were housed in cages until tests and handled daily with free access to food and water and 12 h/12 light/dark cycle. The humidity and temperature were under control. The Shahid Beheshti and Mashhad University of Medical Sciences Animal Ethics Committee approved all animal manipulations.
Surgery and microinjection
Rat anesthetization was done using intraperitoneal combination injection of 100 mg/kg ketamine hydrochloride and 20 mg/kg xylazine. Rats also received penicillin (1.5 × 10
5 U/rat) before placement in a stereotaxic device (Stoelting, USA). The surgical area was cleaned and dried. A drop of dental hemo stop was employed to decrease bleeding. Aβ1-42 was intra-hippocampally (IH) injected. For intra-hippocampal injection, stainless steel guide cannula was placed at the CA1 area of the hippocampus. Stereotaxic coordinates for the dorsal hippocampus were taken from the atlas of Paxinus and Watson (anterior-posterior, 3.8 mm; lateral, ±2.2 mm from the central line, and ventral, 2.7 down from the top of the skull) (
28). Beta-amyloid or PBS (0.5 µL per site = 10 µg) was infused (0.5 mL/min). A Gas Tight Hamilton syringe with Teflon plunger stop (250 μL) was used for the microinjection technique and after each injection, the needle was left in the tissue for 2 min. Penicillin was administered daily, and the rats were allowed 21 days to recover from surgery and after that, behavioral testing (Morris water maze) was performed.
Experimental design
In the present study, the animals were divided into the following groups (n = 7 for each group): 1) control group: had no surgical or dietary intervention. 2) Sham group: injected with the same volume of PBS (0.5 μL per side, IH). 3) Pretreatment with crocin group: received crocin (30 mg/kg, IP) daily in seven days before Aβ1-42 administration (0.1 µg/µL, 0.5 μL per side, IH). 4) Post-treatment with crocin group: received crocin (30 mg/kg, IP) daily in seven days after Aβ1-42 administration (0.1 µg/µL, 0.5 μL per side, IH). 5) Crocin group: only received crocin (30 mg/kg, IP) daily in seven days. 6) Aβ1-42 group: Just received Aβ1-42 (0.1 µg/mL, 0.5 μL per side, IH). Crocin dosage was selected based on the most effective dose of previous studies (
24) (
Scheme 1).
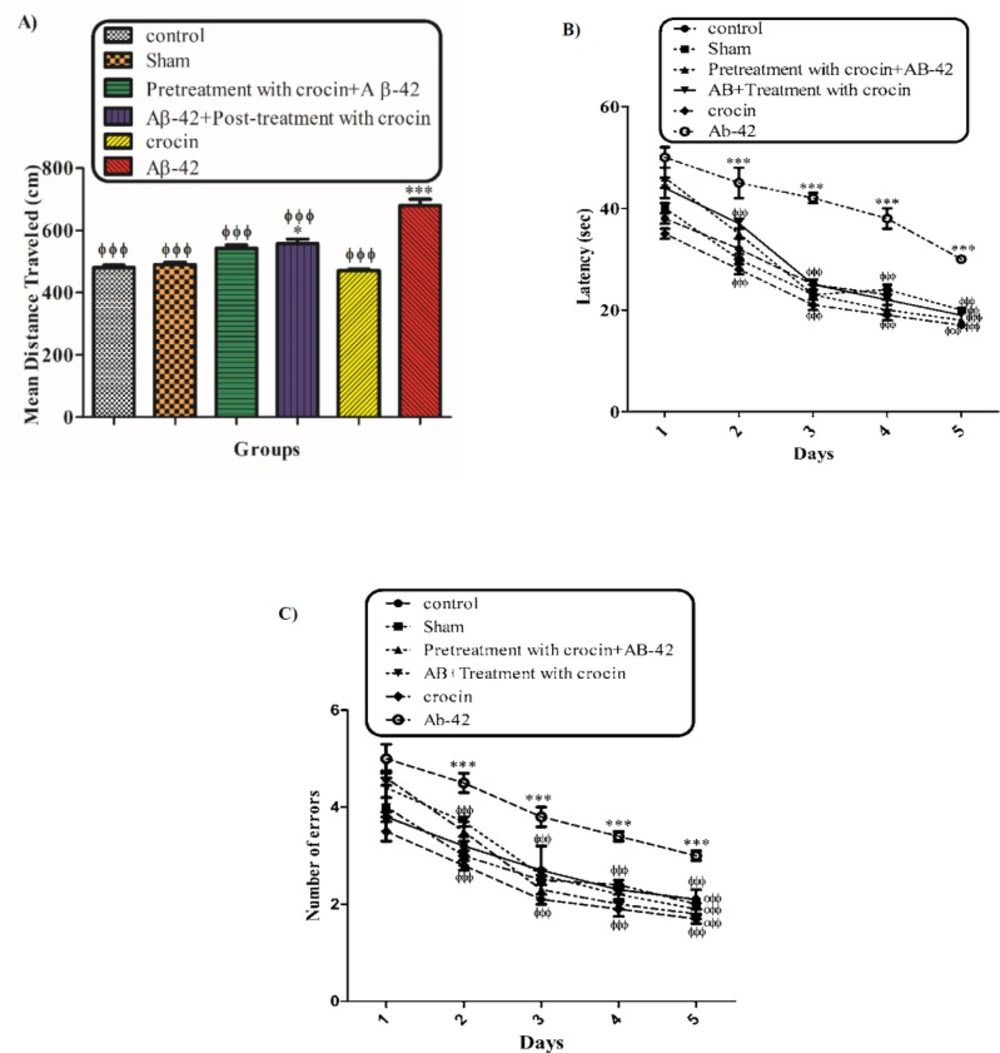
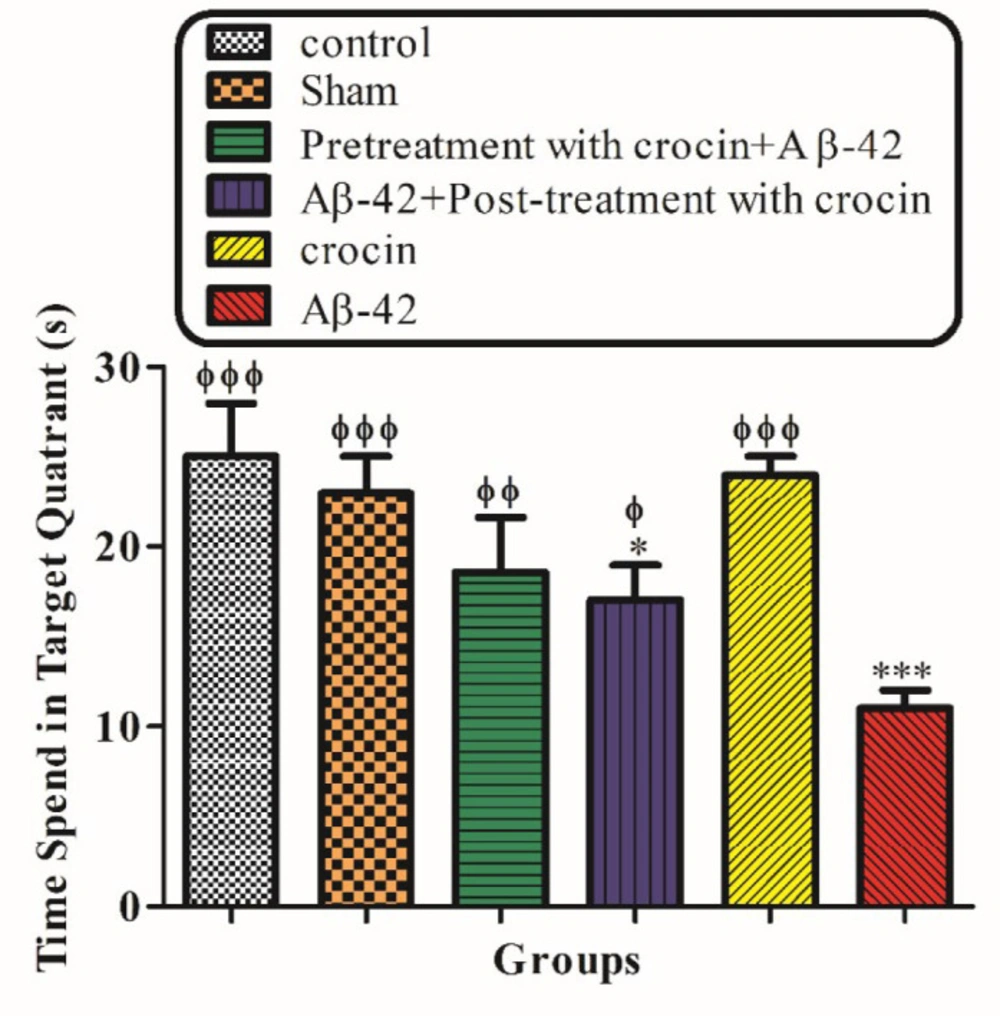
Appraisement of spatial learning and memory by Morris water maze (MWM)
The behavioral tests were performed 21 days after Aβ1-42 injection to assess spatial learning and memory of all the experimental groups. This maze included a black circular pool with a diameter of about 136 cm and a depth of 40 cm. The pool was filled with water to a depth of 35 cm (25 ± 2 °C). This black circular pool was divided into four equal quadrants. The target quadrant (north-west) has an invisible platform in its center that was 10 cm in diameter, which is located 1 cm under the water surface. Rat training was done for 4 days (one block consisting of four trials). In each trial, the rats were randomly placed in one of the four quadrants. A video camera was located just above the pool, linked to a computer and equipped with Ethovision software (Noldus Information Technology, Wageningen, Netherlands) that recorded the swimming pathway and related data. Animals were allowed to swim in the pool for a maximum of 90 s. If the rats did not find the hidden platform within this period, the researcher manually guided the animal to the platform. The rats were then rested on the platform for 20 s. Learning capabilities were measured by quantitative computer data in terms of escape latency (time to find the platform), traveled distance (path length to reach the platform), and swimming speed. After four training days, the rats were tested for probe trials. In the probe trial test, the platform in the target quadrant was removed. The rats were released on the opposite side of the target quadrant and freely swim for 90 s. The time each animal spent in the target quadrant was measured in the probe test (
29).
Evaluation of hippocampal mitochondrial function
Hippocampal mitochondrial isolation
A day after completing MWM, all animals were sacrificed by cervical decapitation, and the hippocampal brain tissues were obtained. Then, with a glass handheld homogenizer, these tissues were minced and homogenized. Mitochondria were prepared from the rats’ hippocampi by differential centrifugation method (
30). First, the samples were centrifuged (1500 g, 10 min, 4 °C), and the broken cell debris and nuclei were sedimented. The supernatant was exposed to centrifugation again (10000 g, 10 min, 4 °C). The upper layer was discarded, and the mitochondrial pellet was washed and suspended in the isolation medium and centrifuged again (10000 ×g, 10 min, 4 °C). Final suspensions of mitochondrial pellets were prepared in Tris buffer containing (0.05 M Tris–HCl, 0.25 M sucrose, 2.0 mMMgCl
2, 20 mMKCl and 1.0 mMNa
2HPO
4, pH 7.4, 4 °C), except for mitochondrial samples used to measure the ROS level, mitochondrial membrane potential (MMP) and swelling, which were incubated in respiration buffer (10 mM Tris, 0.32 mM sucrose, 20 mM Mops, 0.5 mM MgCl
2, 50 mM EGTA, 5 mM sodium succinate and 0.1 mM KH
2PO
4), MMP assay buffer (68 mM D-mannitol, 220 mM sucrose, 10 mMKCl, 5 mMKH
2PO
4, 2 mM MgCl
2, 5 mM sodium succinate, 50 mM EGTA, 10 mM HEPES and 2 mM rotenone) and swelling buffer (3 mM HEPES, 70 mM sucrose, 230 mM mannitol, 5 mM succinate, 2 mMTris-phosphate and 1 mM of rotenone), respectively. Protein concentrations were measured using the Coomassie-blue protein-binding protocol as explained by Bradford (
31). For the normalization process in all the following assays, the mitochondrial samples (0.5 mg mitochondrial protein per ml) were used. All the steps were operated on ice to guarantee high-quality mitochondrial preparation..
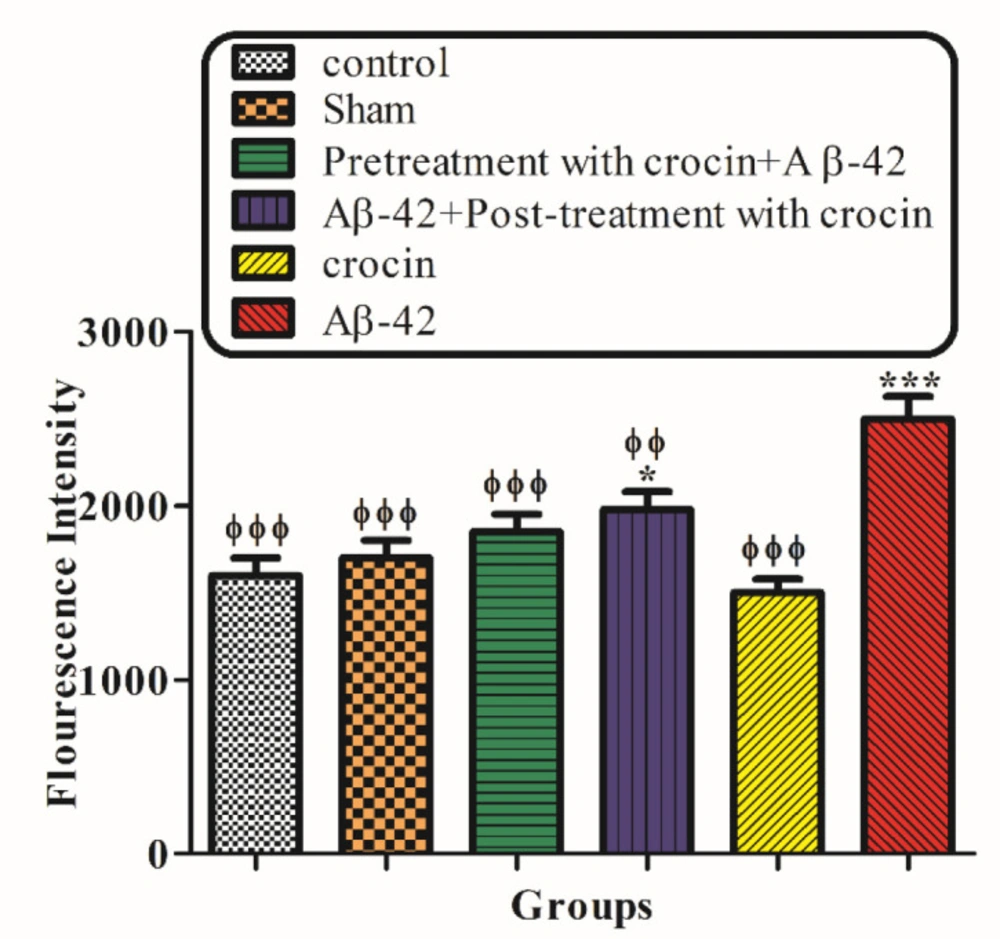
Measurement of the hippocampal mitochondrial ROS levels
The measurements of the mitochondrial ROS levels were performed by a fluorescence spectrophotometer using DCFH-DA. Briefly, isolated mitochondria were incubated with a respiration buffer (
32), and DCFH-DA was added (final concentration, 10 mM) to the mitochondrial samples and then incubated for 10 min. After entry, DCFH-DA was hydrolyzed to non-fluorescent dichlorofluorescein (DCFH), which reacted with ROS and made highly fluorescent dichlorofluorescein (DCF). Then, the fluorescence intensity of DCF was quantified at 60 min using a Shimadzu RF5000U fluorescence spectrophotometer device at excitation and emission wavelengths of 500 nm and 520 nm, respectively (
33).
Measurement of the hippocampal MMP
For the estimation of the MMP, the mitochondrial uptake of a cationic fluorescent dye, Rhodamine 123 (Rh123), was used. Mitochondrial fractions in the MMP assay buffer were incubated with 10 mM of Rh123. Then, the fluorescence was monitored at 60 min using the Shimadzu RF5000U fluorescence spectrophotometer device at an excitation wavelength of 490 and an emission wavelength of 535 nm (
34).
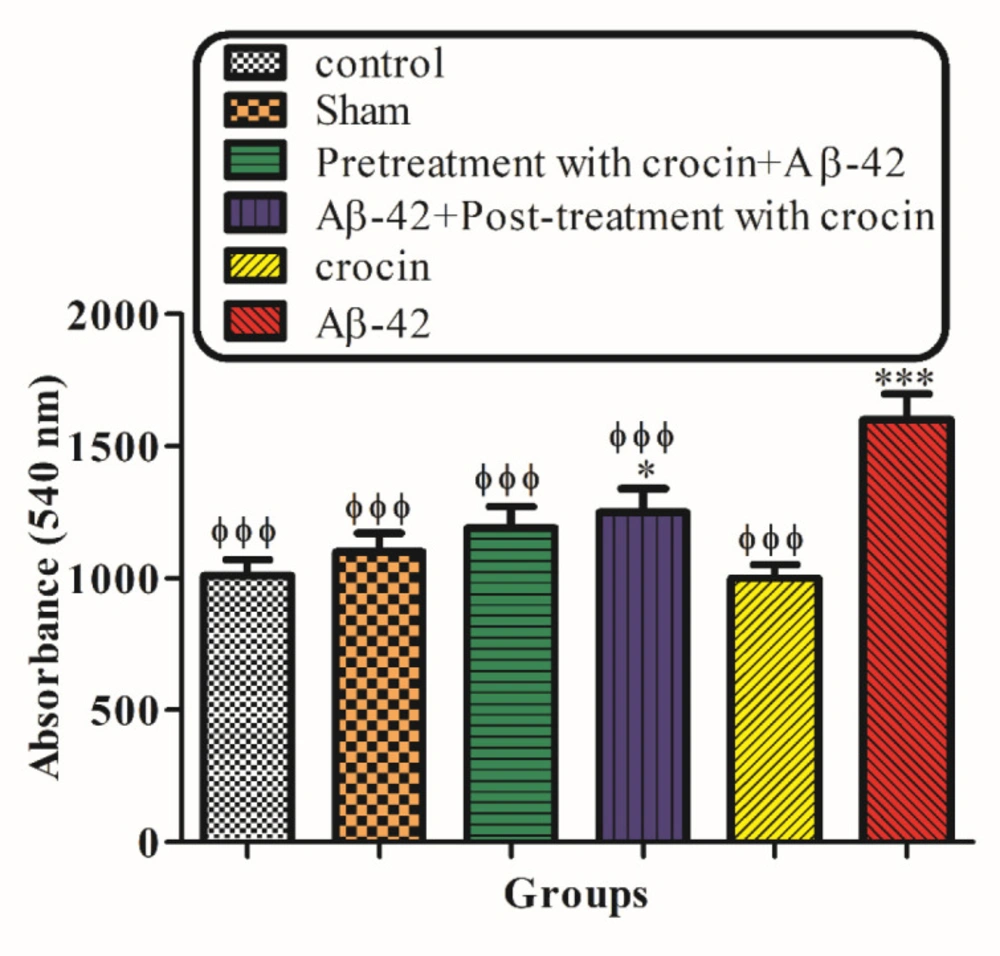
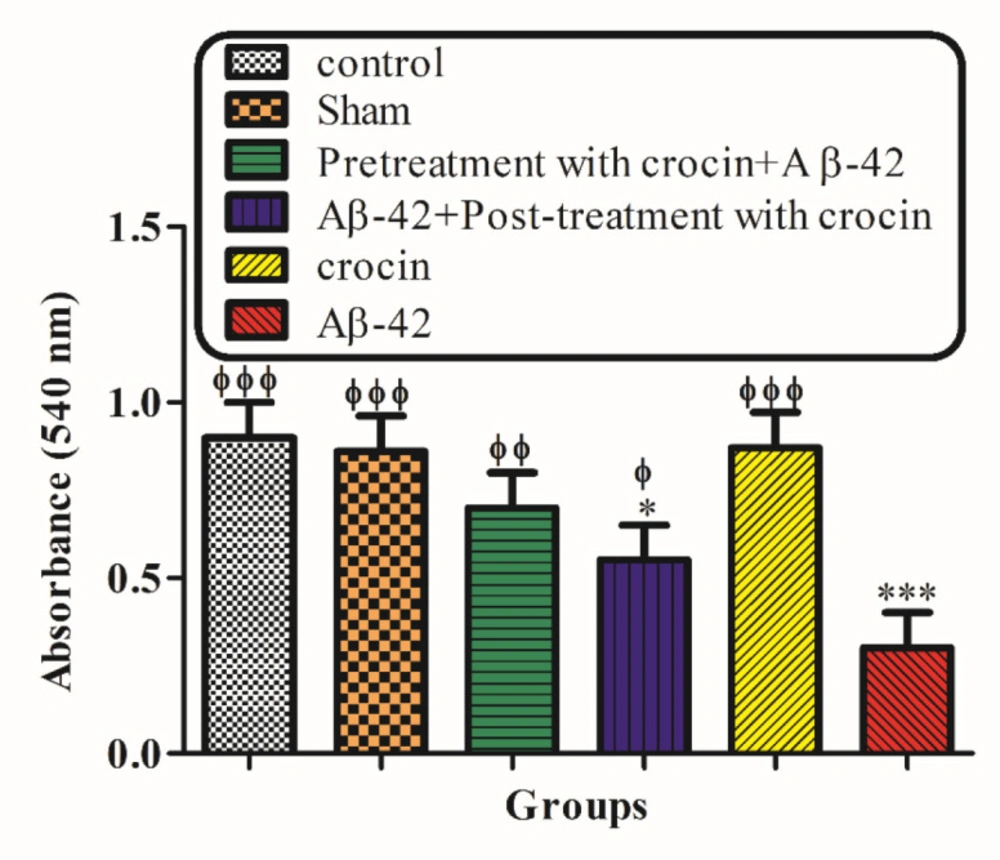
Measurement of hippocampal mitochondrial swelling
The determination of hippocampal mitochondrial swelling was done through changes in light scattering measured spectrophotometrically at 540 nm (30 °C) (
35). The isolated brain mitochondria were suspended in swelling buffer, and the absorbance was determined at 540 nm at 60 min with an ELISA reader apparatus (Tecan, Rainbow Thermo, and Austria). A reduction in absorbance was considered as an indicator of mitochondrial swelling.
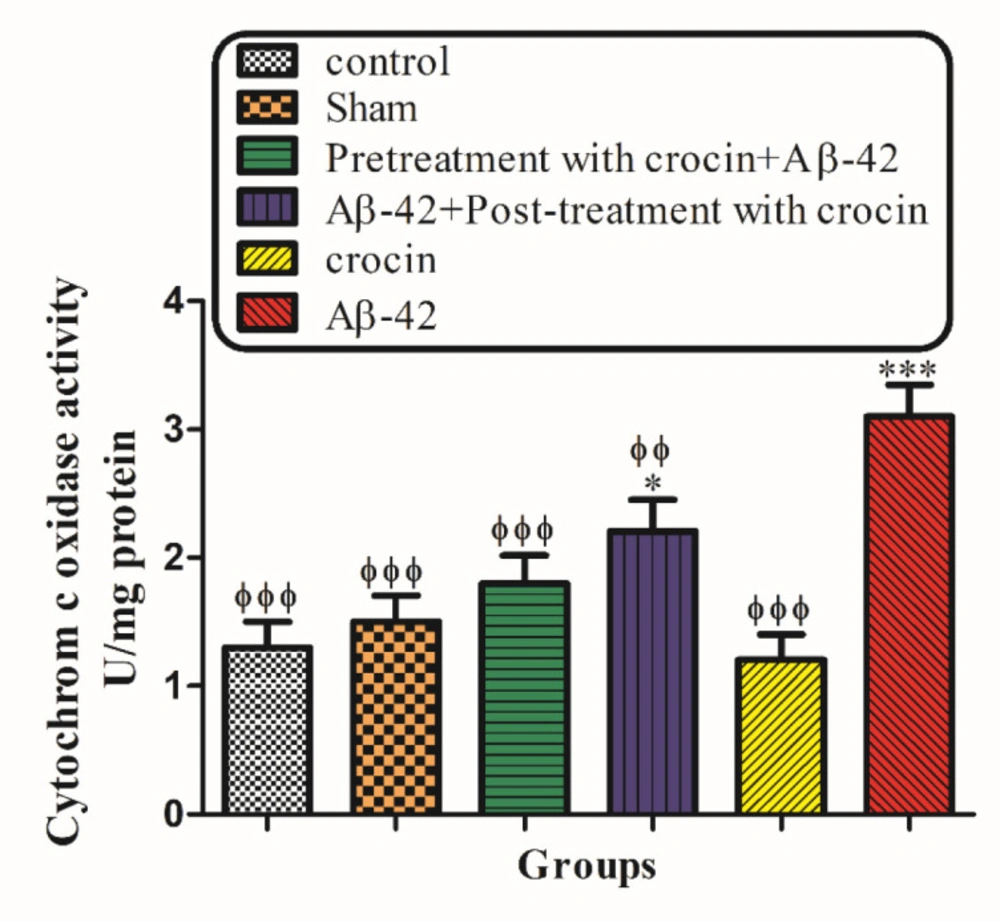
Measurement of cytochrome c oxidase activity and assessment of outer mitochondrial membrane damage
Both mitochondrial outer membrane integrity and cytochrome c oxidase activity were measured using a cytochrome c oxidase assay kit (Sigma, St. Louis, MO). In the colorimetric trial, a decrease in absorbance was caused by oxidation of ferrocytochrome c at 550 nm to ferricytochrome c by cytochrome c oxidase. Experimental procedures were done according to the manufacturer’s protocol; 20 mg of isolated mitochondrial fraction were used for each reaction, and duplicate reactions were performed for each assay. For the determination of total mitochondrial cytochrome c oxidase activity, hippocampal mitochondrial fractions were diluted in the enzyme dilution buffer (10 mMTris–HCl, pH 7.0, containing 250 mM sucrose) with 1 mM n-dodecyl b-ᴅ-maltoside and placed on ice for 30 min. The reaction was performed by adding a fresh ferrocytochrome c substrate solution (0.22 mM) to the sample. The decrease in absorbance at 550 nm is linked to the oxidation of ferrocytochrome c by cytochrome-c oxidase. Cytochrome c oxidase activities were measured and normalized for protein per reaction, and the results were shown as units per milligram of mitochondrial protein. The mitochondrial outer membrane integrity was determined by evaluating the cytochrome-c oxidase activity of the mitochondria in the presence or absence of n-dodecyl b-ᴅ-maltosideas a detergent. The mitochondrial outer membrane damage was measured through the ratio between cytochrome-c oxidase activity eboth in the presence of detergent and its absence (
35).
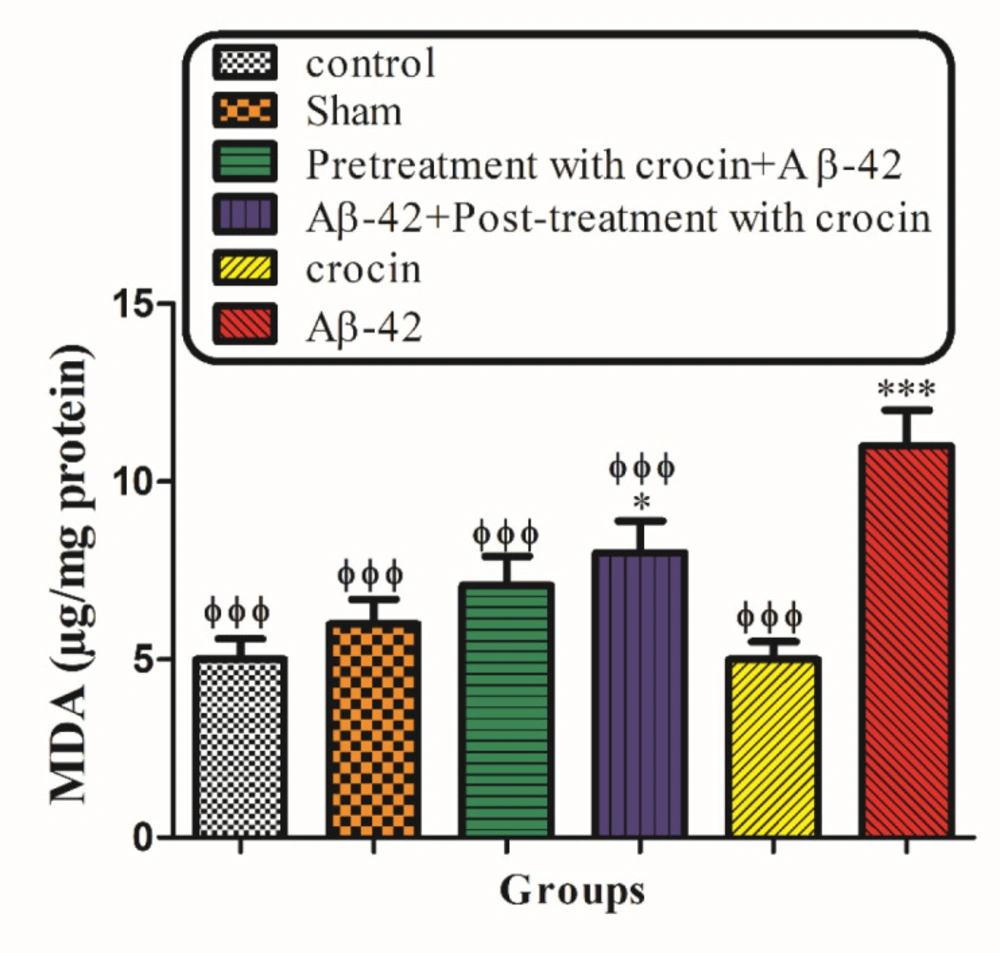
Measurement of mitochondrial lipid peroxidation
The MDA content was measured using the method of Zhang
et al. (
36). The mitochondrial fractions were incubated (1 h) with various concentrations of uranyl acetate at 30 °C; afterward, 0.25 mL sulfuric acid (0.05 M) was added to 0.2 mL mitochondrial fractions, with the addition of 0.3 mL TBA 0.2%. All the microtubes were put in a bath of boiling water for 30 min. Finally, the tubes were placed in an ice bath, and 0.4 mL n-butanol was added to each of them. Then, they were centrifuged (3500 g, 10 min). The amount of MDA formed in each sample was evaluated by measuring the absorbance of the supernatant at 532 nm with an ELISA reader apparatus (Tecan, Rainbow Thermo, Austria). Standard 1,1,3,3-Tetramethoxypropane (TEP) was used, and the MDA content was represented as µg/mg protein (
36).
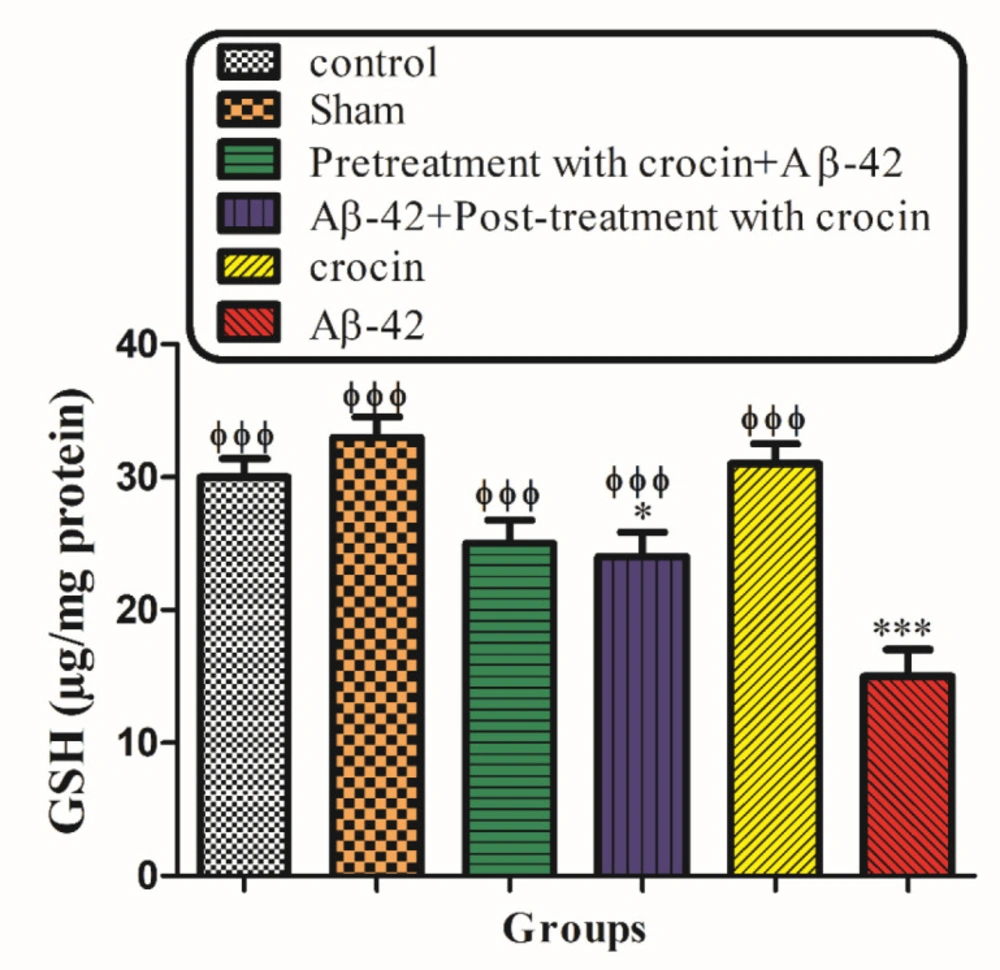
Measurement of mitochondrial Glutathione (GSH) contents
Mitochondrial GSH contents were evaluated using the spectrophotometer method and DTNB as the indicator for the isolated hippocampal mitochondria. The mitochondrial fractions were incubated with various concentrations of uranyl acetate (1 h) at 30 °C. Then the mitochondrial fractions (0.1 mL) were added into phosphate buffers (0.1 mol/L) and DTNB (0.04%) in a total volume of 3.0 mL (pH 7.4). At 412 nm, the yellow color was read on a spectrophotometer (UV-1601 PC, Shimadzu, Japan). The GSH amount was illustrated as µg/mg protein (
37).
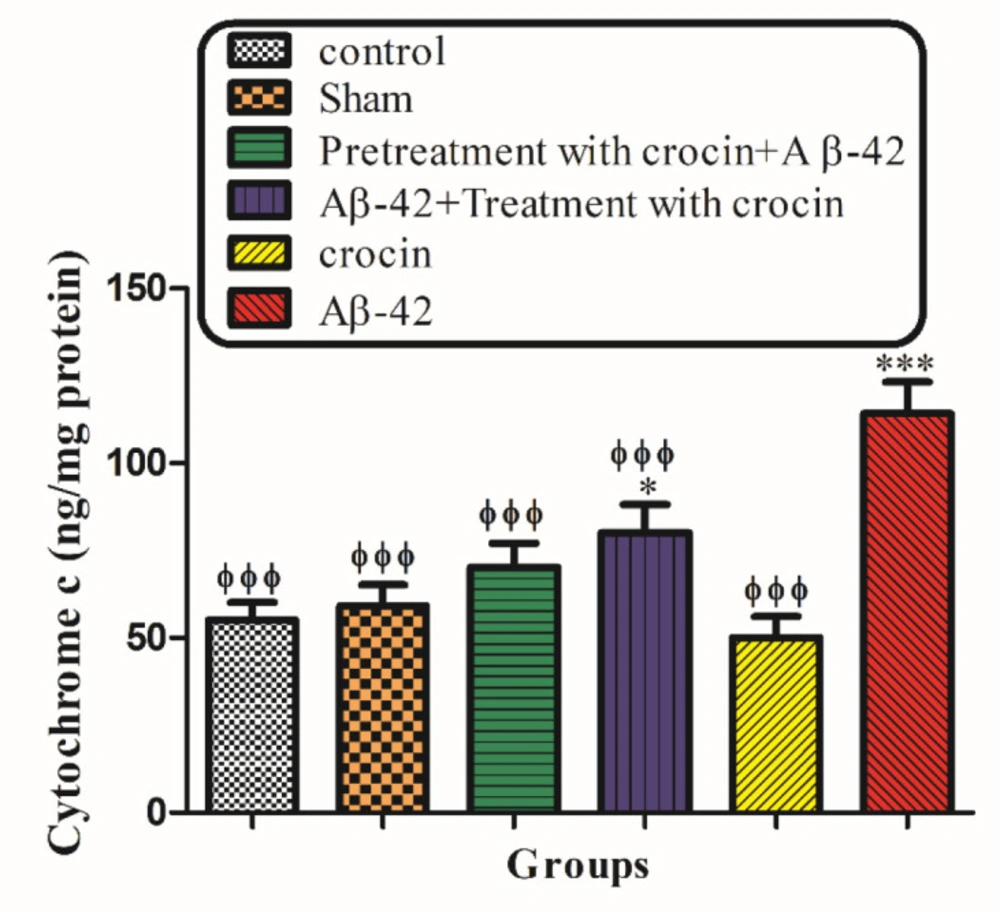
Measurement of mitochondrial Cytochrome c Release
The concentration of cytochrome-c was determined using the QAsntikine Rat/Mouse Cytochrome-c Immunoassay kit provided by R & D Systems, Inc. (Minneapolis, Minn) and the optical density of each well was determined using a spectrophotometer set to 450 nm (
38).
Statistical analysis
The results for each group are presented as mean ± SD. GraphPad Prism-6 (GraphPad Software, La Jolla, CA) was used for the statistical analysis. A mean value for each dependent parameter of memory performance (traveled distance and escape latency) was evaluated over four trials in four training days. Mean values for each dependent measure of mitochondrial function were also calculated just a one-time point (1 h).
Statistical significance between the groups was determined by one-way analysis of variance (ANOVA) using a Bonferroni post hoc multiple comparison test. The P-value was set lower than 0.05.